










| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website http://www.wjon.org |
Case Report
Volume 8, Number 2, April 2017, pages 58-61
Repeated Multiple Neurofibromatosis Type 1 in the Right Lower Limb: A Case Report
Leitao Huanga, b, d, Yi Dinga, b, d, Lai Qia, b, Xia Wuc, Wei Lia, b, Gendong Huanga, b, Min Daia, b, Bing Zhanga, b, e
aDepartment of Orthopedics, Artificial Joints Engineering and Technology, Research Center of Jiangxi Province, Nanchang, China
bDepartment of Orthopedics, The First Affiliated Hospital of Nanchang University, Nanchang, China
cDepartment of College of Pharmacy, Nanchang University, Nanchang 330006, China
dThese authors contributed equally to this work.
eCorresponding Author: Bing Zhang, Department of Orthopedics, The First Affiliated Hospital of Nanchang University, No. 17 Yong Wai Zheng Street, Nanchang 330006, Jiangxi, China
Manuscript accepted for publication April 18, 2017
Short title: Repeated Multiple NF1 in Lower Limb
doi: https://doi.org/10.14740/wjon1011w
This article has been retracted.
| Abstract | ▴Top |
Neurofibromatosis type 1 (NF1) is an autosomal-dominant genetic disease characterized by the presence of multiple neurofibromas. We encountered a unique case of NF1 that manifested as a recurrent soft tissue neurofibroma in the right lower limb that developed over a period of 16 years. The patient presented with a painless mass that was initially diagnosed as inflammatory changes via computed tomography and magnetic resonance imaging. However, the condition was subsequently diagnosed as an intraneural neurofibroma via pathological and immunohistochemical examination, which showed a focal to patchy lymphocytic chronic inflammatory infiltrate and several non-encapsulated masses with clear boundaries that were easily distinguishable from the adjacent neurofibroma. The mass relapsed three times over 3 years since it was discovered, for which the patient underwent comprehensive and complete local resection several times. Postoperative continuous follow-up confirmed that the patient recovered well. Early and complete surgical resection is an effective method for treating and preventing recurrent neurofibromas. However, because of the importance of pathologic examination in the diagnosis of such cases, this uncommon entity might be underreported in patients with NF1.
Keywords: Neurofibroma; Right leg; Fibromatosis; Surgery
| Introduction | ▴Top |
Neurofibromatosis (NF), first described by Von Recklinghausen in 1882 [1], is an autosomal-dominant genetic disease that causes the proliferation of neural crest cells and affects multiple organ systems. NF is characterized by the presence of multiple neurofibromas, cafe-au-lait spots, iris hamartomas (Lisch nodules), and axillary and inguinal freckling [2-4]. Neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2) are classified according to their clinical features and gene mapping characteristics. The incidence of tumors is higher among patients with NF1 than the normal population; moreover, certain types of carcinomas, including breast cancer, may occur more frequently in patients with NF1 [3, 5-7]. Herein, we report a unique case of NF1 wherein multiple recurrent systemic tumors were detected, which could have been easily misdiagnosed if not for the unique pathologic characteristics.
| Case Report | ▴Top |
A 38-year-old man presented to the Department of Orthopedic Oncology of the First Affiliated Hospital of Nanchang University (Nanchang, China) with the chief complaint of a painless mass in the right lower limb that developed over a period of 16 years. The patient appeared projections without apparent inducement at the medial cutaneous right leg which was about 2 × 2 cm in size, fixed block, soft, clear boundary, all directions and passive activities were unlimited at the beginning. Physical examination did not reveal any abnormalities, and the results of routine laboratory tests, such as erythrocyte sedimentation rate, were within the normal range. Three years after the mass was first recognized, a biopsy conducted at the Jian City People’s Hospital, Jiangxi, China showed signs of NF. However, the patient did not report any obvious discomfort, and therefore, the physicians did not recommend further examination but only regular follow-up instead. By early 2012, the patient started to limp. The symptom relieved after walking for a long period; however, the patient subsequently experienced numbness over the skin of the right lower extremity. On July 9, 2013, the patient underwent debridement at a local hospital, wherein the pathology results showed inflammatory cell infiltrations along with ulcers. The treatment did not illicit a favorable response and the tumors were not completely removed. Multiple tumors extending from the right leg to the popliteal fossa, buttocks, and groin were observed, with the largest tumor measuring approximately 10 × 12 cm; knee function was acceptable and no localized tenderness was detected. On April 30, 2014, the patient underwent multiple suction lumpectomies. Postoperative pathological findings indicated multiple schwannomas. However, the patient was hospitalized again owing to a right thigh and groin mass that enlarged; the patient also reported discomfort and weakness in the right calf, and underwent multisite lumpectomy for the right lower limb. The intraoperative findings included irregular shaped masses in the nerves of the right hip, iliac spine, and right lower limb. The masses had a clear boundary, with no obvious signs of invasions to the surrounding tissue.
Computed tomography (CT) revealed several unequally sized lobulated and cystic-solid masses below the right popliteal muscle, with the largest mass measuring 3.7 × 4.5 cm; the masses exhibited a homogeneous density and a clear border (Fig. 1a). No bone hyperplasia or destruction or hardening of the right femur and tibia and fibula were detected; however, enhancement was detected at the edge, a part of the calcaneal bone, and formation of depression. Thus, a diagnosis of borderline or poorly differentiated malignant fibromatosis was established by a professional radiologist.
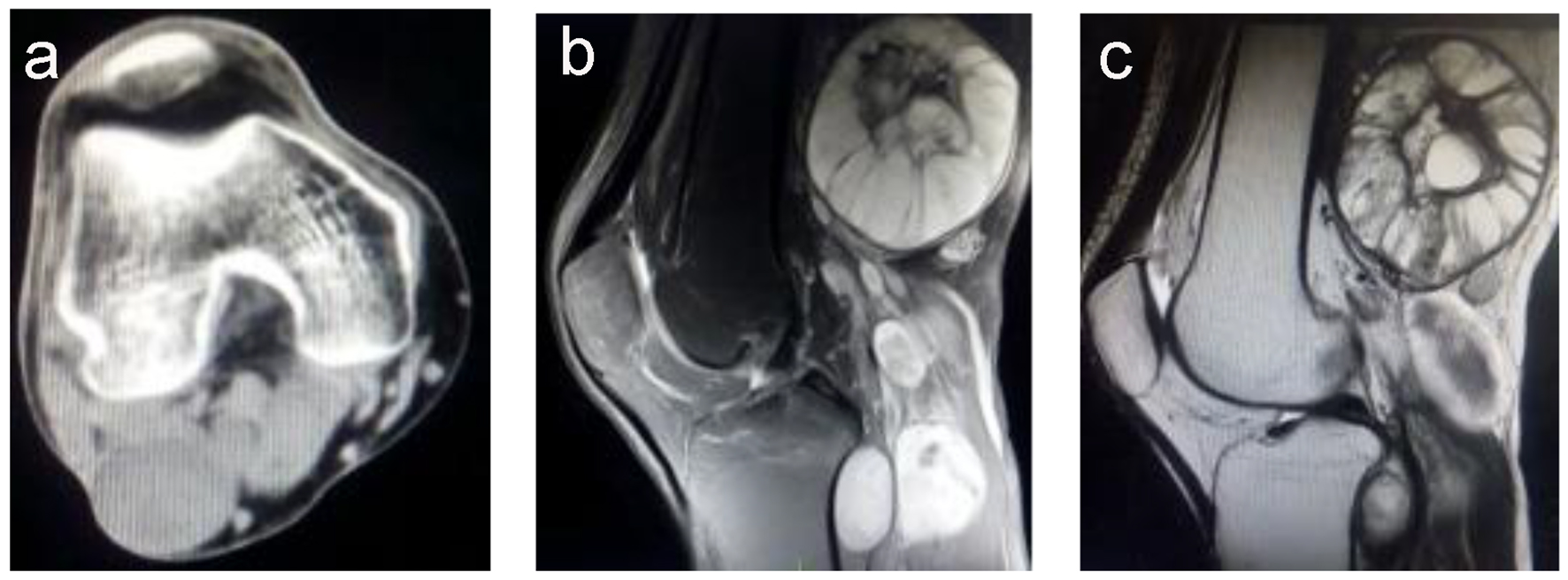 Click for large image | Figure 1. (a) A computed tomography image showing multiple masses with homogeneous density and no bone destruction, with a maximum size of 7.0 × 6.1 cm. (b) A T2-weighted magnetic resonance imaging scan showing a higher signal intensity, with the mass showing an uneven signal and well-defined margins. (c) A T1-weighted magnetic resonance imaging scan showing a slightly higher signal intensity, with the mass showing an uneven signal and liquefaction at the center. |
Magnetic resonance imaging (MRI) showed multiple nodules of varying sizes, mass shadows, and border finishing at the hip, iliac spine, and right tibia and fibula; T2-weighted imaging showed a slightly higher signal intensity (Fig. 1b), and T1-weighted imaging showed a low signal intensity (Fig. 1c). Fat suppression sequence images showed a slightly higher intensity signal; the masses had uneven signals with the largest detected in the right popliteal fossa measuring approximately 7.0 × 6.1 cm2. The tibia and fibula signals were uniform. The tumor borders were well-defined. The soft tissue surrounding the masses demonstrated normal signal intensity. No enlarged lymph nodes or distant metastases were identified. The MRI findings suggested multiple neurofibromas. Therefore, the patient was diagnosed with NF1 and indicated for surgery to resect the tumors.
For the surgery, the patient was placed in a supine position, with disinfected and paved sterile drapes arranged around the right lower limb, iliac spine, groin, and right hip to expose the surgical field following successful epidural anesthesia. First, a medial approach was used to access the inner border of the femoral condyle via an approximately 8 cm long incision. The skin, subcutaneous tissue, superficial fascia, deep fascia, and popliteus were incised to isolate the tumor, which was almost entirely surrounded by a soft tissue capsule. The same process was followed for the masses located at the other sites until all masses were completely excised. The intraoperative findings included multiple irregular but oval masses in the nerves of the right hip, iliac spine, groin, popliteal fossa, calf, and foot. The masses had a clear boundary and no obvious invasion of the surrounding tissue. The nerve membranes were incised to completely remove the tumors.
Gross pathological examination revealed approximately 30 soft, smooth-surfaced, dark gray, oval nodules of varying sizes, with the largest measuring 7.0 × 6.5 × 4.5 cm3. A partially cut section showed a capsule diameter of 0.8 - 1.8 cm containing clear liquid (Fig. 2a). Upon hematoxylin and eosin staining, the membrane-like manifestations were found to be mature connective tissue. The tumor cells were deeply stained and appeared to be spindle-shaped with oval nuclei as well as wavy or irregular in shape, existing in bundles or a swirl or fence-like arrangement. Portions of the tumors showed bleeding, cystic degeneration, and necrosis (Fig. 2b). The neurofibroma component was composed of proliferating spindle cells containing bland cigar-shaped nuclei with inconspicuous nucleoli and an eosinophilic cytoplasm (Fig. 2c). Immunohistochemically, the spindle cells were diffusely positive for S-100 protein (3+) and the Ki-67 labeling index was < 2%. Moreover, the tumor was partially positive for CD34, CD56, and CR but negative for NF. Taken together, the surgical findings, tumor location, and nodule pathology resulted in a diagnosis of primary NF1. There was no evidence of NF transformation.
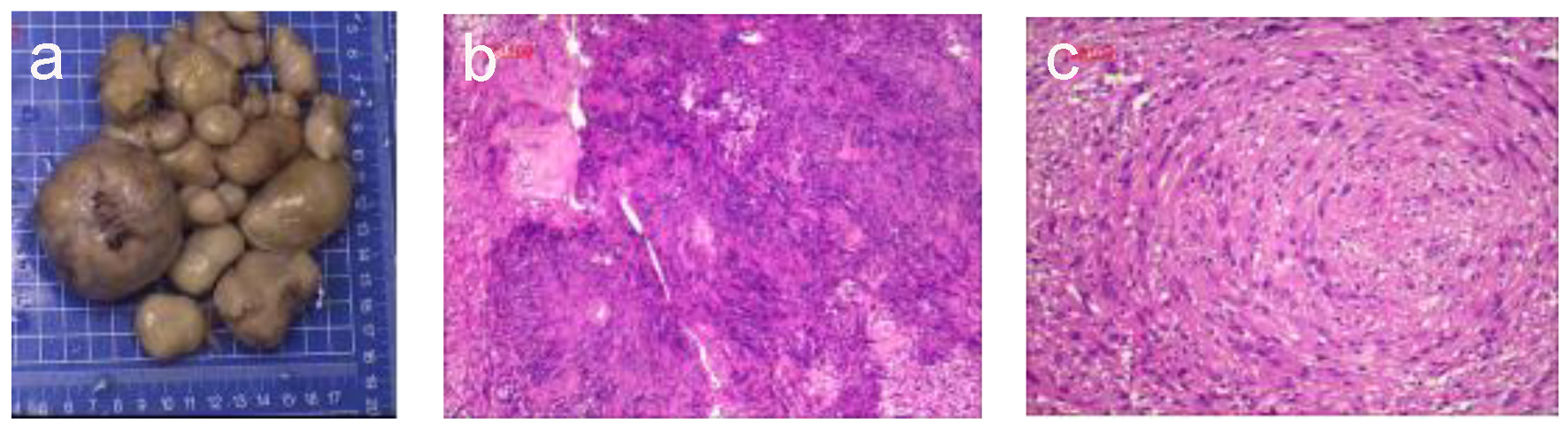 Click for large image | Figure 2. Histological observations. (a) The largest excised specimen was dark gray and measured 7 × 6.5 × 4.5 cm. (b) Photomicrograph of the neurofibroma component showed papillary proliferation of cells with cystic degeneration and a lobular arrangement (hematoxylin-eosin staining; magnification, × 100). (c) The neurofibroma was composed of proliferating spindle cells containing bland cigar-shaped nuclei with inconspicuous nucleoli and an eosinophilic cytoplasm. The lobules in the masses comprised a tight swirl of cells, though the focal areas demonstrated closely clustered thin-spindled and stellate cells (hematoxylin-eosin staining; × 100). |
The patient was discharged without any complications 1 week after the total excision surgery. At the 6-month follow-up, the patient was completely free from pain during daily activities and demonstrated normal range of motion in the right hip and knee joint. No evidence of recurrence was noted. This study has been approved by the Ethical Advisory Committee of The First Affiliated Hospital of Nanchang University Medical School (Nanchang, Jiangxi, China), and informed consent was obtained from the patient for publication of this case report and any accompanying images.
| Discussion | ▴Top |
NF1 is an autosomal-dominant inherited disorder with an estimated worldwide incidence of approximately 1:3,000 individuals, which makes NF1 the most common hereditary multitumor syndrome [8-10]. Jokinen et al examined the clinicopathologic and immunohistochemical features of 11 derm, and showed that superficial atypical NF is morphologically unusual but has no apparent association with NF1 or a short-term risk of recurrence or malignant transformation [11]. A distinctive histologic feature observed in the neurofibroma component of the present case was the focal to patchy lymphocytic chronic inflammatory infiltrate, which probably represents a marker for this type of neurofibroma. Neurofibromas are non-encapsulated and usually involve multiple lesions in the presence or absence of NF1. Neurilemmomas, on the other hand, are solitary encapsulated tumors, typically attached to or surrounded by a nerve and appear to push axons aside [12, 13]. The term neurofibroma has been used synonymously with neurilemmoma (neurinoma, neuroma, schwannoma, perineural fibroma, and peripheral glioma) [14]. However, in our case, the multiple masses were sharply demarcated and easily distinguishable from the adjacent neurofibroma.
Patients with NF1 are at an increased risk for several non-epithelial neoplasms, including neurofibromas, malignant peripheral nerve sheath tumors, gliomas, leukemia, pheochromocytoma, and rhabdomyosarcoma [4, 5]. Furthermore, studies have also suggested an increased risk for certain types of carcinomas in patients with NF1 [5, 6, 7]. NF1 is caused by inactivating mutations in the NF1 gene, which is located on chromosome 17q11.2 [8, 9]. This gene has a tumor suppressor function as the gene product of NF1, neurofibromin, is a major negative regulator of the RAS/mitogen-activated protein kinase pathway, which transmits mitogenic signals to the nucleus [15].
The genetic mechanism of carcinoma development in patients with NF1 is not well understood. However, Guran and Safali [16] reported a case of breast carcinoma in an NF1 patient wherein loss of heterozygosity of the NF1 gene was observed in the carcinoma tissue; such findings support the role of the NF1 gene in the etiology of some cases of breast cancer. Biallelic NF1 inactivation is observed in neurofibromas and malignant peripheral nerve sheath tumors [17-19]. Astrocytomas in patients with NF1 also demonstrate a loss of neurofibromin expression and a loss of heterozygosity of the NF1 gene [20]. Furthermore, previous studies claim that the loss of heterozygosity of the NF1 region has been observed in the majority of pheochromocytoma cases in patients with NF1 [21].
In the present case, a 38-year-old man with a 16-year clinical history of recurrent masses was diagnosed with neurofibroma via imaging and pathological examination. CT and MRI identified the neoplasm between the deep fascia and the muscle. However, in the present case, the mass had relapsed three times over 3 years, except upon its discovery. Based on the unique clinical characteristics and the presence of multiple masses of different sizes, to the best of our knowledge, the current case report represents the first description of NF1 with specific and untypical biological behaviors, which occurred over a prolonged period and involved relapse. In patients with NF1, a positive family history is reported in approximately 50% of cases [9]. However, in our case, the patient demonstrated a negative family history, and the NF1 could have been misdiagnosed if not for the distinguished pathological characteristics. The clinical manifestations, imaging findings, intraoperative and postoperative findings together resulted in the diagnosis of NF1. NF1 involves unilateral disease in > 80% of cases and bilateral or adrenal disease in 10% of cases. Immunohistochemically, NF1 typically demonstrates negative results for the S-100 protein [8, 22]. Thus, NF1 requires a differential diagnosis that includes multiple tumors of the small intestine, metastatic hematologic tumor (such as melanoma), polyp syndrome (such as Peutz-Jeghers syndrome), gastrointestinal stromal tumor, carcinoid tumor, adrenal tumor, adrenal adenoma, pheochromocytoma, and paraganglioma benign neurofibroma. The most commonly affected site of NF1 is the digestive tract. Diverse clinical symptoms are commonly observed in gastrointestinal NF and gastrointestinal stromal tumors [4, 5].
Following surgical treatment, the patient was symptom-free at the 6-month follow-up. Although he remains well and without abnormalities, owing to the risk for recurrence and additional risk for malignancy, long-term follow-up should be continued, with particular attention to signs of malignancy [23]. In conclusion, the present case highlights the rare manifestation of NF1 as a soft tissue neurofibroma. This uncommon entity might be underreported in patients with NF1.
| References | ▴Top |
- Reynolds RM, Browning GG, Nawroz I, Campbell IW. Von Recklinghausen's neurofibromatosis: neurofibromatosis type 1. Lancet. 2003;361(9368):1552-1554.
doi - Brems H, Beert E, de Ravel T, Legius E. Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol. 2009;10(5):508-515.
doi - Ishida M, Okabe H. Cutaneous squamous cell carcinoma in a patient with neurofibromatosis type 1: A case report. Oncol Lett. 2013;6(4):878-880.
doi - Zoller ME, Rembeck B, Oden A, Samuelsson M, Angervall L. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer. 1997;79(11):2125-2131.
doi - Seminog OO, Goldacre MJ. Risk of benign tumours of nervous system, and of malignant neoplasms, in people with neurofibromatosis: population-based record-linkage study. Br J Cancer. 2013;108(1):193-198.
doi pubmed - Salemis NS, Nakos G, Sambaziotis D, Gourgiotis S. Breast cancer associated with type 1 neurofibromatosis. Breast Cancer. 2010;17(4):306-309.
doi pubmed - Sharif S, Moran A, Huson SM, Iddenden R, Shenton A, Howard E, Evans DG. Women with neurofibromatosis 1 are at a moderately increased risk of developing breast cancer and should be considered for early screening. J Med Genet. 2007;44(8):481-484.
doi pubmed - Schaefer IM, Strobel P, Thiha A, Sohns JM, Muhlfeld C, Kuffer S, Felmerer G, et al. Soft tissue perineurioma and other unusual tumors in a patient with neurofibromatosis type 1. Int J Clin Exp Pathol. 2013;6(12):3003-3008.
pubmed - Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123(1):124-133.
doi pubmed - Agaimy A, Vassos N, Croner RS. Gastrointestinal manifestations of neurofibromatosis type 1 (Recklinghausen's disease): clinicopathological spectrum with pathogenetic considerations. Int J Clin Exp Pathol. 2012;5(9):852-862.
pubmed - Jokinen CH, Argenyi ZB. Atypical neurofibroma of the skin and subcutaneous tissue: clinicopathologic analysis of 11 cases. J Cutan Pathol. 2010;37(1):35-42.
doi pubmed - Sullivan MJ, Babyak JW, Kartush JM. Intraparotid facial nerve neurofibroma. Laryngoscope. 1987;97(2):219-223.
doi pubmed - Neely JG. Neoplastic involvement of the facial nerve. Otolaryngol Clin North Am. 1974;7(2):385-396.
pubmed - Sellars SL. Neurofibroma of the facial nerve. S Afr Med J. 1972;46(37):1332-1334.
pubmed - Seizinger BR. NF1: a prevalent cause of tumorigenesis in human cancers? Nat Genet. 1993;3(2):97-99.
doi pubmed - Guran S, Safali M. A case of neurofibromatosis and breast cancer: loss of heterozygosity of NF1 in breast cancer. Cancer Genet Cytogenet. 2005;156(1):86-88.
doi pubmed - Laycock-van Spyk S, Thomas N, Cooper DN, Upadhyaya M. Neurofibromatosis type 1-associated tumours: their somatic mutational spectrum and pathogenesis. Hum Genomics. 2011;5(6):623-690.
doi pubmed - Garcia-Linares C, Fernandez-Rodriguez J, Terribas E, Mercade J, Pros E, Benito L, Benavente Y, et al. Dissecting loss of heterozygosity (LOH) in neurofibromatosis type 1-associated neurofibromas: Importance of copy neutral LOH. Hum Mutat. 2011;32(1):78-90.
doi pubmed - Upadhyaya M, Kluwe L, Spurlock G, Monem B, Majounie E, Mantripragada K, Ruggieri M, et al. Germline and somatic NF1 gene mutation spectrum in NF1-associated malignant peripheral nerve sheath tumors (MPNSTs). Hum Mutat. 2008;29(1):74-82.
doi pubmed - Gutmann DH, Donahoe J, Brown T, James CD, Perry A. Loss of neurofibromatosis 1 (NF1) gene expression in NF1-associated pilocytic astrocytomas. Neuropathol Appl Neurobiol. 2000;26(4):361-367.
doi pubmed - Bausch B, Borozdin W, Mautner VF, Hoffmann MM, Boehm D, Robledo M, Cascon A, et al. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with pheochromocytoma and neurofibromatosis type 1. J Clin Endocrinol Metab. 2007;92(7):2784-2792.
doi pubmed - Hornick JL, Fletcher CD. Soft tissue perineurioma: clinicopathologic analysis of 81 cases including those with atypical histologic features. Am J Surg Pathol. 2005;29(7):845-858.
doi - Yu X, Guo R, Fan C, Liu H, Zhang B, Nie T, Tu YI, et al. Aneurysmal bone cyst secondary to a giant cell tumor of the patella: A case report. Oncol Lett. 2016;11(2):1481-1485.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.