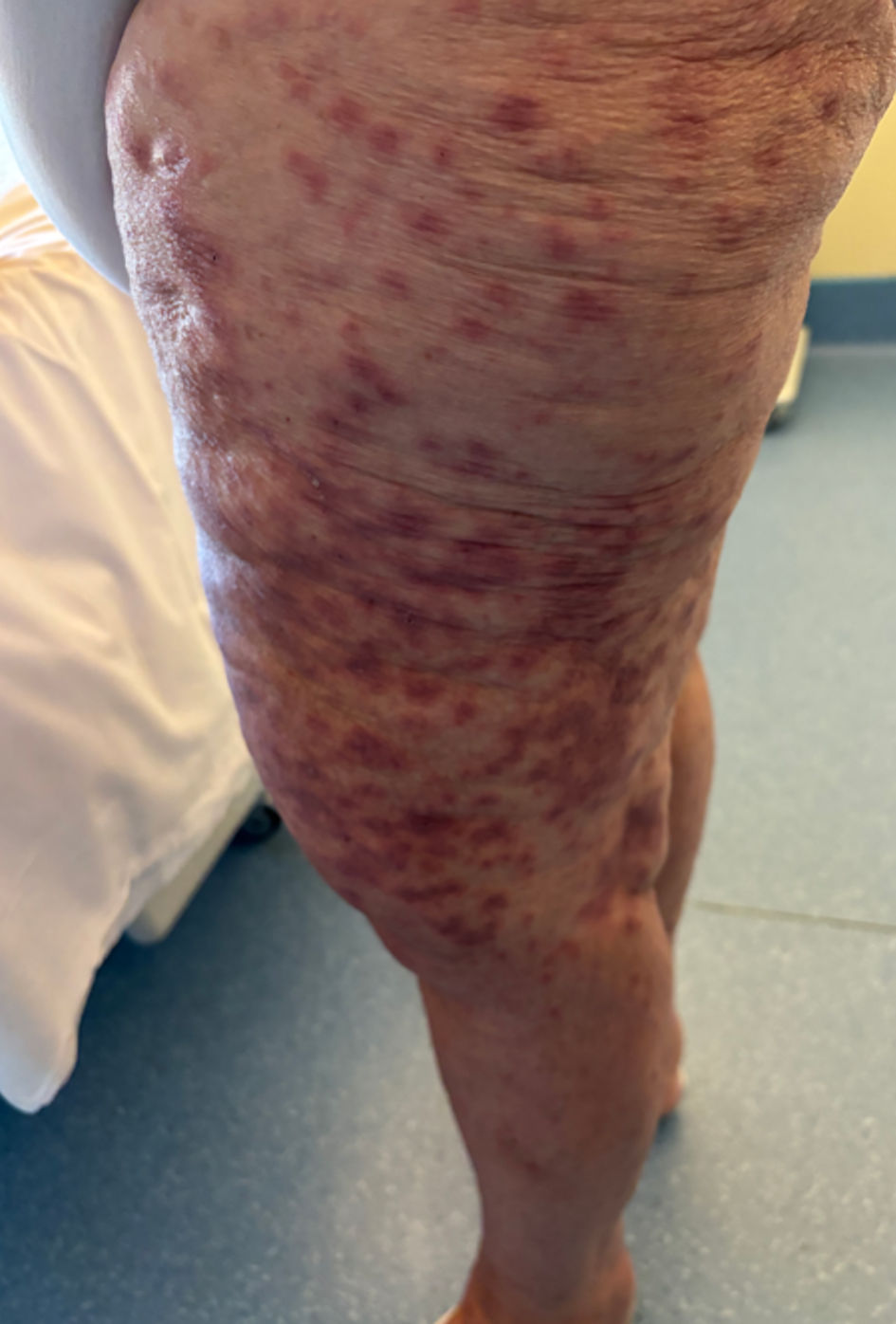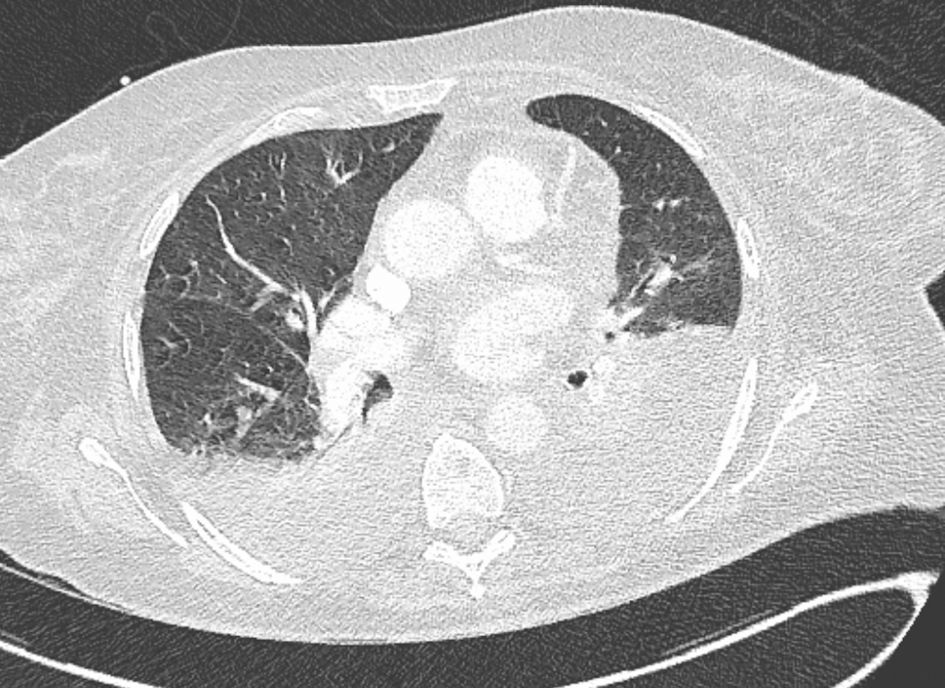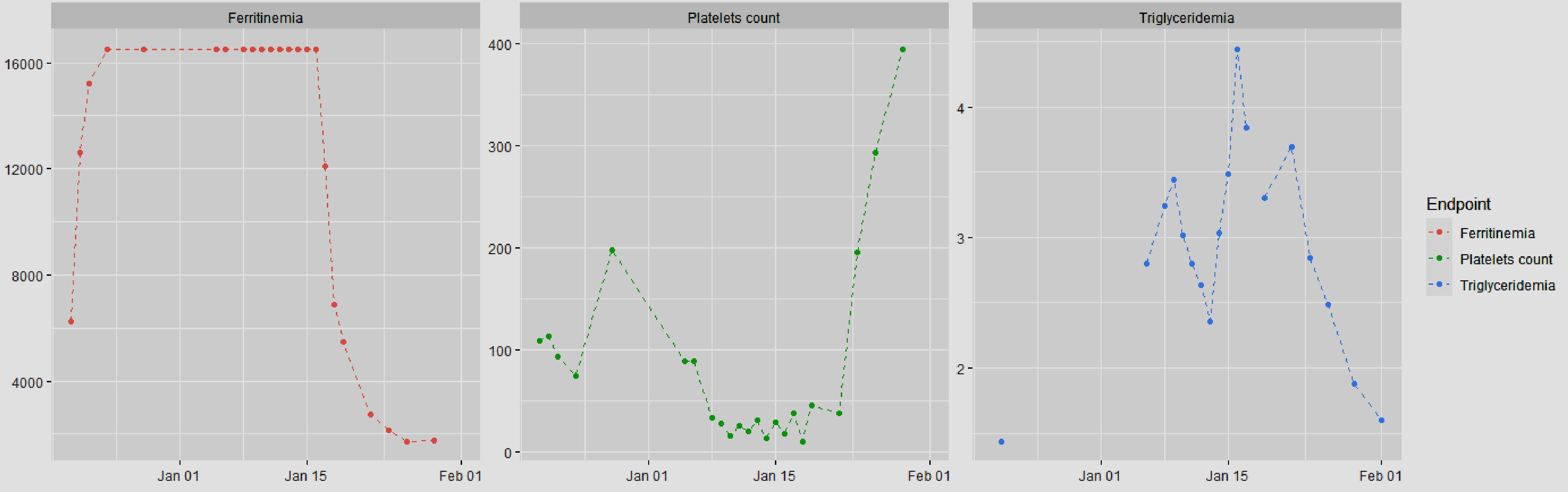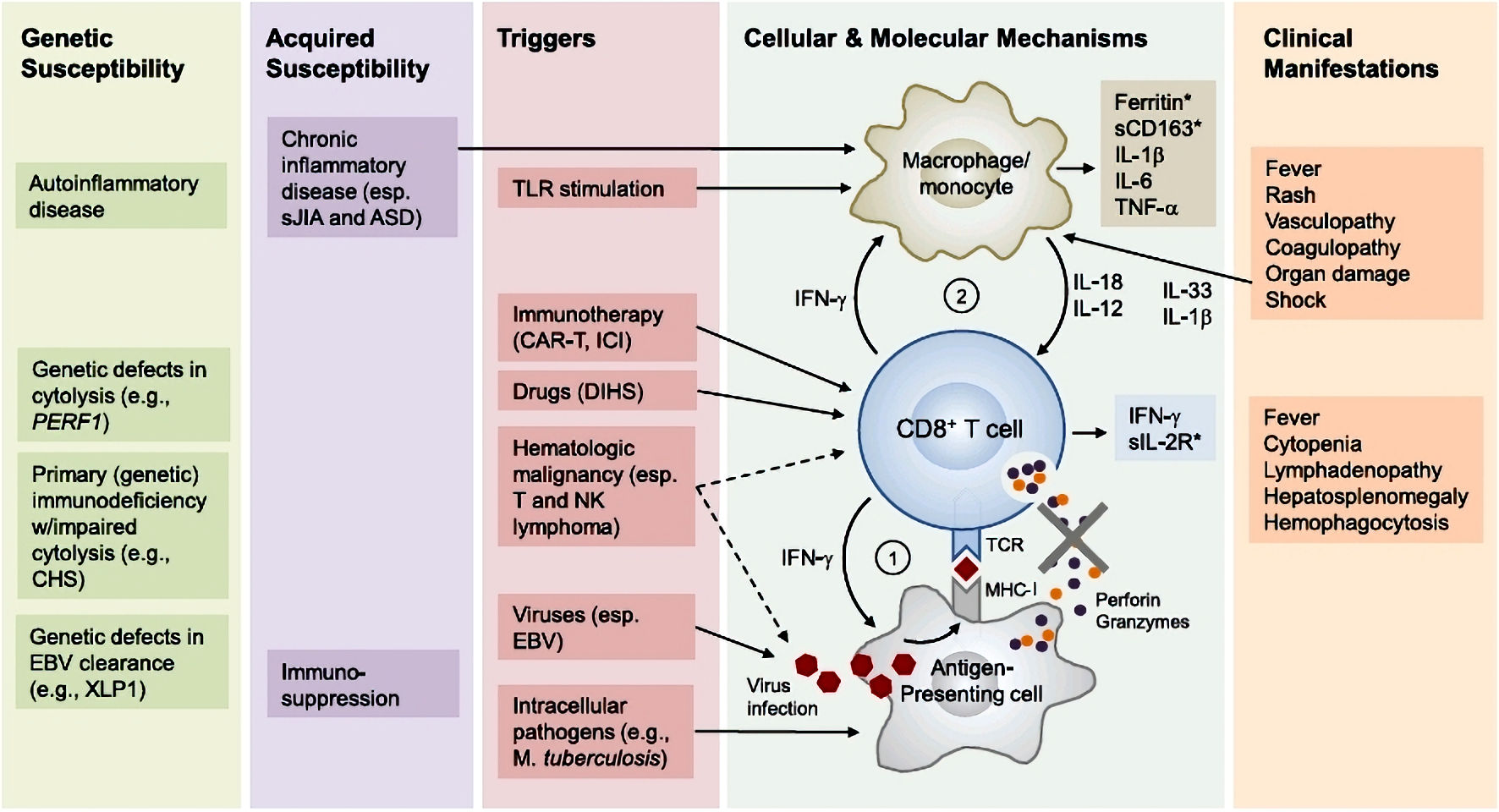
Figure 1. Pathophysiology of hemophagocytic lymphohistiocytosis (adapted from [2]). Primary HLH involves susceptibility genes necessary for the functioning of the immune system, whether it be related to autoinflammatory diseases or a deficiency in clearing certain viruses. Additionally, acquired susceptibility can play a role. The secondary mechanism is triggered by factors such as a viral (EBV) or bacterial (Mycobacterium tuberculosis) infection, the use of an immune checkpoint inhibitor, or a malignant hematopathy. Subsequently, there is an inflammation cascade involving antigen-presenting cells and inflammatory cytokines such as IFN-γ or IL-12, leading to hyperactivation of cytotoxic T lymphocytes and macrophages. This hyperactivation is responsible for the various described symptoms. Notably, the hyperactivation of immune cells involves the tyrosine kinases JAK 1 and 2 at the intracellular level. ASD: autoimmune systemic disease; CART: chimeric antigen receptor T-cell therapy; CD163: cluster of differentiation 163; CHS: Chediak-Higashi syndrome; DHS: drug hypersensitivity syndrome; EBV: Epstein-Barr virus; ICI: immune checkpoint inhibitor; IFN-γ: interferon gamma; IL: interleukin; MHC-I: major histocompatibility complex class I; NK: natural killer; sIL-2R: soluble interleukin-2 receptor; TCR: T-cell receptor; TLR: Toll-like receptor; TNF-α: tumor necrosis factor alpha; XLP1: X-linked lymphoproliferative disease 1.