

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website http://www.wjon.org |
Case Report
Volume 9, Number 4, August 2018, pages 123-127
A Case of Recurrent Pregnancy-Induced Adult Onset Familial Hemophagocytic Lymphohistiocytosis
Lan Y. Wanga, John Hub, Giridharan Ramsinghb, Bassam Theodoryb, Bassam Yaghmourc, Maria Vergara-Llurid, George Yaghmourb, e
aDepartment of Medicine, University of Southern California, Keck School of Medicine of USC, Los Angeles, CA 90033, USA
bJane Anne Nohl Division of Hematology and Center for the Study of Blood Diseases, University of Southern California, Keck School of Medicine of USC, USC Norris Comprehensive Cancer Center, Los Angeles, CA 90033, USA
cDepartment of Pulmonary and Critical Care, University of Southern California, Keck School of Medicine of USC, Los Angeles, CA 90033, USA
dDepartment of Pathology, University of Southern California, Keck School of Medicine of USC, Los Angeles, CA 90033, USA
eCorresponding Author: George Yaghmour, University of Southern California, NTT3467, 1441 Eastlake Avenue, Los Angeles, CA 90033, USA
Manuscript submitted August 19, 2018, accepted August 27, 2018
Short title: Pregnancy-Induced Adult Onset Familial HLH
doi: https://doi.org/10.14740/wjon1145w
| Abstract | ▴Top |
Hemophagocytic lymphohistiocytosis (HLH) is a rare and potentially fatal disease primarily of children, characterized by a severe hyperinflammatory state. We describe a case of adult onset familial HLH with a novel exon 19, c.1607G>T (p.Arg536Leu) heterozygous mutation of the UNC13D gene in a 40-year-old woman who developed HLH during her first and second pregnancies, both episodes occurring during the first trimester. Our patient was treated successfully both times with HLH-94 protocol following spontaneous abortions and is currently in the process of getting a bone marrow transplant. We also discuss pregnancy as a potential trigger for late onset familial HLH.
Keywords: Hemophagocytic lymphohistiocytosis; Pregnancy; Familial; Adult; Mutation; Chemotherapy; Genetic
| Introduction | ▴Top |
Hemophagocytic lymphohistiocytosis (HLH) is a rare and life-threatening entity characterized by uncontrolled inflammation due to the abnormal inability of cytotoxic T lymphocytes and natural killer (NK) cells to downregulate macrophages, resulting in exuberant macrophage activation. The excess cytokine generation leads to the clinical manifestations of the disease, which include persistent fevers, hepatosplenomegaly and cytopenias, and hemophagocytic phenomena in the bone marrow, lymph node, liver and spleen. The disease is highly fatal and prompt diagnosis and treatment are crucial, but its under-recognition due to its rarity and complex clinical presentation often leads to delayed or missed diagnosis, contributing to its high mortality [1]. The annual incidence worldwide is estimated at one per 800,000 with the majority of adult cases located in Asia [2].
The disease is divided into familial HLH (FHL) and secondary HLH. In the FHL type, patients have a genetic predisposition to developing the disease [3]. Defects in the autosomal recessive genes PRF1 (FHL2), UNC13D (FHL3), STX11 (FHL4), and STXBP2 (FHL5) have been implicated in the development FHL. These genes are involved in the cytolytic secretory pathway and are important in delivering preformed granules such as granzymes and perforins to the target cells of T lymphocytes and NK cells [3, 4]. The frequency of these mutations is dependent on an individual’s ethnicity with the UNC13D mutation as the most common in the Korean population [5, 6]. In contrast, acquired, or secondary forms of HLH most commonly occur due to extreme immunologic activation, which can be triggered by infections, autoimmune disorders, and malignancy, with leukemia and lymphoma being the most common associated neoplasms [7, 8]. Retrospective analysis of 276 adult patients hospitalized for secondary HLH looking at novel predictors of mortality, showed that malignancy-associated secondary HLH, older age, and worse renal function were associated with worse outcomes, and that outcomes were predicated on the extent of organ dysfunction at diagnosis [9]. The onset of secondary HLH has also been reported to occur during pregnancy, typically after the second trimester [10-12]. The variable immunologic disturbances and shifts in response to existence of foreign genetic fetal material during pregnancy are thought to produce a favorable scenario to triggering HLH [10, 11].
The onset of FHL is typically in infancy and early childhood, but on rare occasion, cases of late-onset FHL have been reported [13]. Here, we report a unique case of adult onset FHL with an associated novel exon 19, c.1607G>T (p.Arg536Leu) heterozygous mutation in UNC13D, which has not yet been reported to cause UNC13D-related disease, and that was potentially triggered twice by pregnancy.
| Case Report | ▴Top |
Our patient is a 40-year old G0P0 Korean woman with no significant past medical or family history, who had initially presented to the emergency department (ED) after 1 week of fevers, shortness of breath and generalized malaise. In the ED, she was found to be unresponsive and in respiratory arrest and shock. Laboratory values were notable for pancytopenia, disseminated intravascular coagulation (DIC), acute liver failure, and positive β-hCG (Table 1). Computed tomography (CT) image of her abdomen showed evidence of non-cirrhotic portal hypertension with diffuse lymphadenopathy and splenomegaly. The patient was subsequently intubated and admitted to the intensive care unit (ICU).
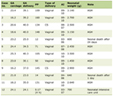 Click to view | Table 1. Labs Upon Presentation, After Treatment, Outpatient Clinic 2 Months Prior to Admission and Upon Re-Presentation |
Hematology was consulted upon admission and bone marrow biopsy was performed. The biopsy displayed a hypercellular bone marrow with evidence of extensive erythrophagocytosis (i.e. macrophages with numerous intracytoplasmic erythrocytes) and hemophagocytosis (i.e. macrophages with intracytoplasmic white blood cells and nuclear debris) in the bone marrow aspirate smears and trephine biopsies (Fig. 1). No malignant cells were seen in the bone marrow biopsy. Based on elevated ferritin levels (4,122 ng/mL), splenomegaly (18 cm), pancytopenia, hypofibrinogenemia (< 60 mg/dL), persistent fever > 38.5 °C, and morphologic evidence of bone marrow hemophagocytosis, she was diagnosed with HLH and initiated on a modified HLH-94 protocol with 75% dose reduction of etoposide given severe mucositis (Table 2). She completed the 8-week regimen with clinical improvement and was discharged to home.
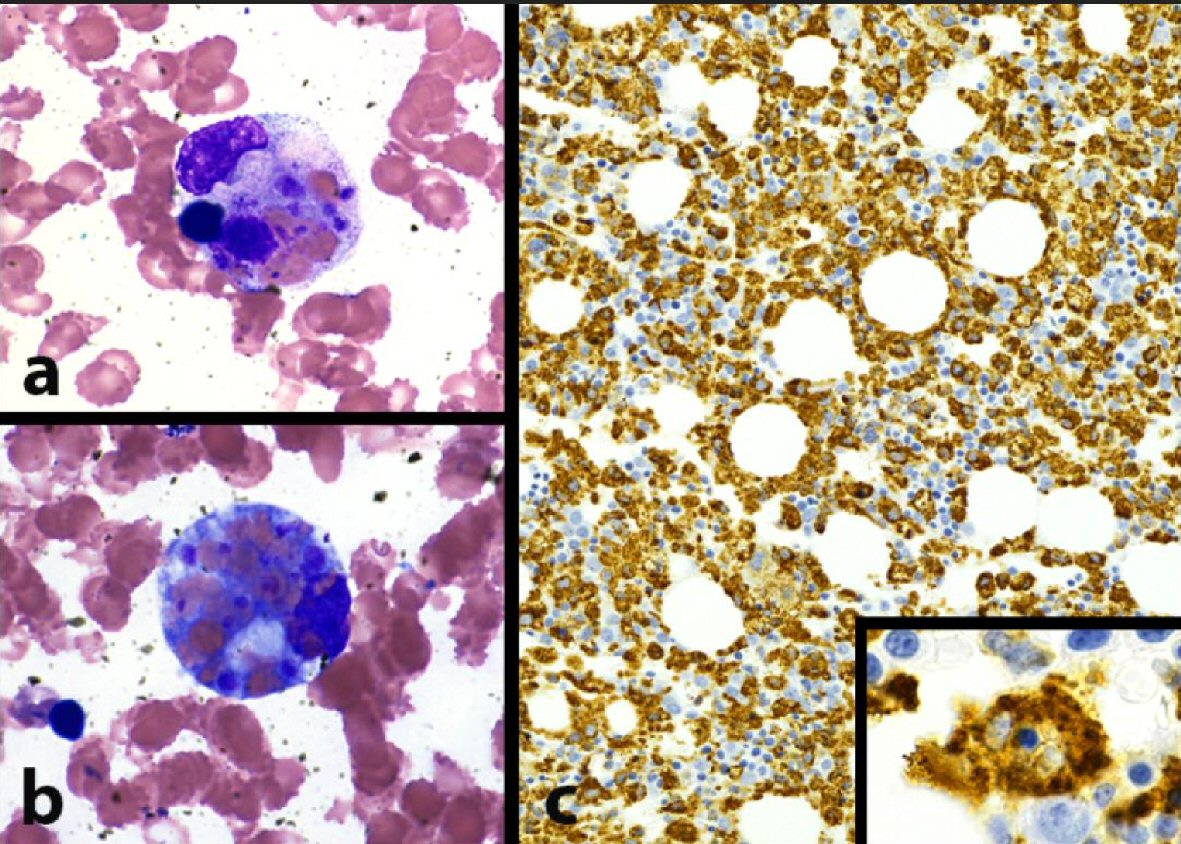 Click for large image | Figure 1. Morphologic evidence of hemophagocytosis: bone marrow pathology pictures. (a) Macrophage with intracytoplasmic nuclear debris, red blood cells, and platelets (hemophagocytosis) (BM aspirate smear, Wright Giemsa stain, 1,000 × magnification). (b) Macrophage with numerous intracytoplasmic red blood cells (erythrophagocytosis) (BM aspirate smear, Wright Giemsa stain, 1,000 × magnification). (c) Extensive macrophage infiltration of bone marrow, highlighting scattered hemophagocytic cells (inset, 1,000 × magnification) (BM trephine core biopsy, CD68 immunohistochemical stain). |
Click to view | Table 2. Modified HLH-94 Protocol |
The etiology of her episode of HLH was unclear. An extensive infectious disease workup was negative, including blood and/or urine bacterial, mycobacterial, and fungal cultures, as well as serologies for HIV, EBV, parvovirus, CMV, HSV, Hepatitis A, B and C, Cryptococcus, coccidioidomycosis, Histoplasma, Bartonella, Brucella, Coxiella, and syphilis were negative. A lymph node needle core biopsy was also performed, which likewise demonstrated morphologic evidence of hemophagocytosis, with sinus histiocytosis showing increased erythrophagocytosis and rare hemophagocytic cells. Special stains (AFB, GMS, and Brown-Hopps) were negative for microorganisms. No malignancy was seen on this needle core biopsy. An extensive rheumatologic workup was also negative, which included ANA, ANCA, actin antibody IgG, dsDNA, SM/RNP, SSA/SSB, Scl-70 Ab, and Jo-1 Ab serologies. In addition, our patient did not meet clinical criteria for adult onset Still’s disease or systemic lupus erythematosus, which are the two most common causes of autoimmune triggers for secondary HLH. Because of her initially elevated β-hCG, the obstetrics and gynecologic (OB/GYN) service evaluated our patient, who ultimately concluded that she had an early pregnancy with subsequent spontaneous abortion. Although pelvic ultrasound was negative, the OB/GYN consultant determined that the pregnancy was likely too early to be seen on imaging and that she had a spontaneous abortion given her downtrending β-hCG levels. Although diffuse lymphadenopathy and splenomegaly were observed on abdominal CT, additional CT imaging of head, chest, abdomen, and pelvis did not show definitive evidence of malignancy.
A 21-gene panel (including ADA, AP3B1, BLOC1S6, BTK, CD27, IL2RA, IL2RG, ITK, LYST, MAGT1, MVK, PNP, PRF1, RAB27A, SH2D1A, SLC7A7, STX11, STXBP2, UNC13D, WAS, and XIAP) was sent to a reference laboratory to test for predisposition mutations for HLH. Our patient was found to be positive for an exon 19, c.1607G>T (p.Arg536Leu) heterozygous mutation of uncertain significance, on the UNC13D gene.
Eight months after her initial diagnosis of HLH, our patient relapsed, presenting with fevers, worsening cytopenias, rising ferritin levels, worsening liver function tests, and coagulopathy. She was also found to have an elevated β-hCG to 53,630 mIU/mL with her pelvic ultrasound showing evidence of an intrauterine pregnancy at 6 weeks of gestation. She was offered an elective abortion by OB/GYN, but developed a spontaneous abortion the day after admission. The patient was then re-initiated on the modified HLH-94 protocol and completed the 8-week course of therapy, with resolution of disease. Given the concern for pregnancies triggering HLH, our patient had subdermal etonogestrel birth control implant device placed.
Due to this presumed genetic predisposition, the decision was made for the patient to be evaluated for allogeneic hematopoietic stem cell transplantation (HSCT). Our patient’s sister was tested with the mutation panel as described above and was negative for mutations. Currently, our patient is completing a transplant workup in preparation for a haploidentical HSCT from her sister.
| Discussion | ▴Top |
Herein, we describe, to our knowledge, the first case of adult onset FHL with an exon 19, c.1607G>T (p.Arg536Leu) heterozygous mutation of uncertain significance, on the UNC13D gene, that was potentially triggered twice by the first trimester pregnancies. This gene sequence change results in replacement of an arginine residue with leucine at codon 536 of the UNC13D protein. The arginine residue is weakly conserved and there is a moderate physicochemical difference between arginine and leucine. This variant is present in population databases (rs143305366, ExAC 0.006%), but up until now, this variant has only been reported in two pediatric HLH cases (a 1-year old female and a 12-year old female) with unclear significance in the pathogenesis of the disease [14]. However, in our patient’s case, this variant may be associated with type 3 FHL.
Our patient developed the disease in adulthood twice in the setting of elevated β-hCG with a confirmed intrauterine pregnancy seen on imaging during her second episode. This unique case suggests that our patient had a genetic predisposition lowering the threshold for developing disease in the setting of an appropriate trigger, such as pregnancy. Notably our patient denied history of pregnancy, severe infections, or other severe illness prior to her initial episode. It is unclear why pregnancy, as opposed to other insults to the immune system, resulted in her developing HLH.
Pregnancy-associated HLH is a rare entity with only about 20 case reports in the literature and typically also associated with infection, malignancy, and rheumatologic disorders. The diagnosis and onset of symptoms are typically manifest during the second trimester, usually resulting in spontaneous abortion, often with improvement in symptoms afterwards. The earliest reported case was at 10 weeks of gestation [10-12, 15-18]. However, in our patient, she presented both times in her first trimester: first, she was diagnosed with HLH at a serum β-hCG level indicative of very early pregnancy (around 3 weeks); on the second she had an HLH flare when she had a confirmed pregnancy at 6 weeks of gestation. The association of HLH symptoms in our patient both times with an elevated β-hCG in the setting of a negative infectious, rheumatologic, and malignancy workup suggests that pregnancy was the inciting factor for her HLH.
The pathogenesis of pregnancy as a trigger for HLH is currently not well understood but multiple theories have been proposed [15-19]. One theory is that the immature placenta releases trophoblastic debris with fetal RNA and DNA components into maternal circulation, leading to a systemic inflammatory response [16, 17]. Osugi et al in 1997 hypothesized that an abnormal increase in Th1/Th2 ratio that can happen during pregnancy leads to macrophage activation and hemophagocytosis [18]. In contrast, Yamaguchi et al in 2005 stated that the shift from a Th1 predominance to Th2 predominance (in order for immune adaptation to the genetically foreign fetus) results in a decreased cell-mediated immunity, allowing for increased susceptibility to infections, and leading to macrophage overactivation as the first immune response to infection [19]. In our patient, we believe her heterozygous UNC13D mutation likely predisposed her to immune dysregulation/overactivation and along with the shift from Th1 to Th2 dominance unique to pregnancy, resulted in the clinical manifestations of adult onset familial HLH.
Management of HLH consists of treating the overactive inflammation, as well as eliminating the underlying cause. Our patient had a spontaneous abortion and was subsequently placed on the HLH-94 protocol during both of her episodes. The only curative therapy for HLH is allogeneic HSCT. Indications for allogeneic HSCT include patients with FHL and those with relapsed disease, like our patient.
In summary, adult onset FHL is becoming better recognized, as in our patient, who was diagnosed at the age 40. While FHL and secondary HLH were previously thought to be distinct entities and mutually exclusive, our case demonstrates the emerging view that the two forms are a continuum rather than separate diseases: her genetic predisposition likely lowering the threshold for developing disease in the setting of an appropriate trigger. It may be reasonable to consider testing more adults for known HLH mutations in the setting of HLH related to pregnancy or in those with an unclear trigger.
Conflict of Interest
Authors have no conflict of interest to declare.
| References | ▴Top |
- Jamy O, Nunnery S, Giri S, Wiedower E, Johnson B, Yaghmour G, Martin MG. Under-recognition of hemophagocytic syndrome in United States' rural, non-teaching hospitals. Leuk Lymphoma. 2016;57(12):2911-2913.
doi pubmed - Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. 2014;383(9927):1503-1516.
doi - Mayson E, Saverimuttu J, Warburton P. Two-faced haemophagocytic lymphohistiocytosis: comparative review of two cases of adult haemophagocytic lymphohistiocytosis. Intern Med J. 2014;44(2):198-201.
doi pubmed - Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, Villanueva J, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood. 2011;118(22):5794-5798.
doi pubmed - Yoon HS, Kim HJ, Yoo KH, Sung KW, Koo HH, Kang HJ, Shin HY, et al. UNC13D is the predominant causative gene with recurrent splicing mutations in Korean patients with familial hemophagocytic lymphohistiocytosis. Haematologica. 2010;95(4):622-626.
doi pubmed - Seo JY, Song JS, Lee KO, Won HH, Kim JW, Kim SH, Lee SH, et al. Founder effects in two predominant intronic mutations of UNC13D, c.118-308C>T and c.754-1G>C underlie the unusual predominance of type 3 familial hemophagocytic lymphohistiocytosis (FHL3) in Korea. Ann Hematol. 2013;92(3):357-364.
doi pubmed - Henter JI, Arico M, Egeler RM, Elinder G, Favara BE, Filipovich AH, Gadner H, et al. HLH-94: a treatment protocol for hemophagocytic lymphohistiocytosis. HLH study Group of the Histiocyte Society. Med Pediatr Oncol. 1997;28(5):342-347.
doi - Fisman DN. Hemophagocytic syndromes and infection. Emerg Infect Dis. 2000;6(6):601-608.
doi pubmed - Johnson B, Giri S, Nunnery SE, Wiedower E, Jamy O, Yaghmour G, Chandler JC, et al. Comorbidities drive outcomes for both malignancy-associated and non-malignancy-associated hemophagocytic syndrome. Clin Lymphoma Myeloma Leuk. 2016;16(4):230-236.
doi pubmed - Rousselin A, Alavi Z, Le Moigne E, Renard S, Tremouilhac C, Delluc A, Merviel P. Hemophagocytic syndrome in pregnancy: case report, diagnosis, treatment, and prognosis. Clin Case Rep. 2017;5(11):1756-1764.
doi pubmed - Rajendran A, Sherif AA, Divakar A, Surendran S, Visalakshy J, Pillai MPG. Triple threat: pregnancy, SLE, EBV as potential triggers in secondary hemophagocytic lymphohistiocytosis. International Journal of Research in Medical Sciences. 2017;5(8):3758.
doi - Alsina L, Colobran R, de Sevilla MF, Catala A, Vinas L, Ricart S, Plaza AM, et al. Novel and atypical splicing mutation in a compound heterozygous UNC13D defect presenting in Familial Hemophagocytic Lymphohistiocytosis triggered by EBV infection. Clin Immunol. 2014;153(2):292-297.
doi pubmed - Ueda I, Kurokawa Y, Koike K, Ito S, Sakata A, Matsumora T, Fukushima T, et al. Late-onset cases of familial hemophagocytic lymphohistiocytosis with missense perforin gene mutations. Am J Hematol. 2007;82(6):427-432.
doi pubmed - Chen X, Wang F, Zhang Y, Teng W, Wang M, Nie D, Zhou X, et al. Genetic variant spectrum in 265 Chinese patients with hemophagocytic lymphohistiocytosis: Molecular analyses of PRF1, UNC13D, STX11, STXBP2, SH2D1A, and XIAP. Clin Genet. 2018;94(2):200-212.
doi pubmed - He M, Jia J, Zhang J, Beejadhursing R, Mwamaka Sharifu L, Yu J, Wang S, et al. Pregnancy-associated hemophagocytic lymphohistiocytosis secondary to NK/T cells lymphoma: A case report and literature review. Medicine (Baltimore). 2017;96(47):e8628.
doi pubmed - Shukla A, Kaur A, Hira HS. Pregnancy induced haemophagocytic syndrome. J Obstet Gynaecol India. 2013;63(3):203-205.
doi pubmed - Teng CL, Hwang GY, Lee BJ, Wang RC, Chou MM. Pregnancy-induced hemophagocytic lymphohistiocytosis combined with autoimmune hemolytic anemia. J Chin Med Assoc. 2009;72(3):156-159.
doi - Osugi Y, Hara J, Tagawa S, Takai K, Hosoi G, Matsuda Y, Ohta H, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood. 1997;89(11):4100-4103.
pubmed - Yamaguchi K, Yamamoto A, Hisano M, Natori M, Murashima A. Herpes simplex virus 2-associated hemophagocytic lymphohistiocytosis in a pregnant patient. Obstet Gynecol. 2005;105(5 Pt 2):1241-1244.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.