

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Letter to the Editor
Volume 12, Number 2-3, June 2021, pages 77-80
Primary Ewing Sarcoma of the Stomach
Kenan Hallona, Subhi Mansoura, Mira Damounya, Amir Faraha, Miriam Obeida, Rozan Marjiyeha, Jawad Ghaliaa, Safi Khuria, b, c
aGeneral Surgery Department, Rambam Health Care Campus, Haifa, Israel
bHPB and Surgical Oncology Unit, Rambam Health Care Campus, Haifa, Israel
cCorresponding Author: Safi Khuri, HPB and Surgical Onclology Unit, General Surgery Department, Rambam Health Care Campus, Haa’leya Hashniya, Haifa 31096, Israel
Manuscript submitted April 10, 2021, accepted April 26, 2021, published online May 14, 2021
Short title: Primary Ewing Sarcoma of the Stomach
doi: https://doi.org/10.14740/wjon1380
| To the Editor | ▴Top |
Ewing sarcoma (ES), a malignant tumor of small round blue cells, arises usually in skeletal tissues and lacks neuroectodermal differentiation [1]. It was reported firstly by James Ewing in 1921. Although previously regarded as a separate entity from primitive neuroectodermal tumor (PNET), which exhibits neuroectodermal features, nowadays, they are treated as the same tumor due to the similarity in chromosomal translocation and immunohistochemical characteristics [1]. These tumors belong to the ES family of tumors which include skeletal ES/PNET, extraskeletal ES/PNET and Askin tumor (thoracopulmonary PNET). These tumors originate from distinctive mesenchymal progenitor cells due to their similar histologic and immunohistochemical characteristics. ES is a highly metastatic type of sarcoma and represents almost 10-15% of bone sarcomas, and is regarded as the second most common primary malignant bone tumor [2]. Although 5-year survival and overall survival have been improved significantly recently, the recurrence rate of such tumors remains high.
| Classification | ▴Top |
ES/PNET tumors can be categorized as skeletal affecting bones and deep soft tissues [3] and extraskeletal affecting other organs. Skeletal ES/PNET represents the most common type at about 80-85% of cases. Extraskeletal organs affected, including the pancreas, small bowel, kidneys, stomach, esophagus, ovaries and vagina, represent almost 15-20% of ES/PNET tumors [4]. Other cohort series including 120 - 2,202 patients reported an incidence rate of 21-31% [5, 6]. Both types usually affect children and young adults, with the extraskeletal type affecting slightly older children than skeletal ES by 5 - 10 years, with an average age of 20 years, thus, granting the extraskeletal subtype particular importance in the adult population. Although skeletal ES/PNET has slight male predilection (M/F ratio = 1.5:1), extraskeletal subtypes have an equal sex predilection (Table 1, [4]). Even though patients suffering extraskeletal ES/PNET are treated by the same protocol of skeletal ES/PNET, there is no consensus on whether the two diseases carry a similar prognosis.
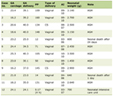 Click to view | Table 1. The Differences Between ES/PNET Subtypes |
| Diagnosis | ▴Top |
Accurate diagnosis of ES/PNET can be made by immunohistochemical staining for different markers which show variable results. Immunohistochemical staining for a monoclonal antibody to cluster of differentiation (CD)99 (HBA/71, 12E7, and 013) is positive in almost all cases of ES/PNET, while immunohistochemical staining for intermediate filament vimentin is usually positive [7, 8]. Immunohistological reagents show variable immunohistochemical staining include synaptophysin, chromogranin A, S100 and neuron-specific enolase [9].
Fluorescent in situ hybridization (FISH) or reverse transcription polymerase chain reaction can also be used for the diagnosis of said tumors, by testing the presence of genetic mutations (11;22) (q24;q12) translocation (EWS/FLI1 fusion) [9]. The previously mentioned genetic mutations are essential criterion for the diagnosis of ES/PNET, although sometimes these tests are negative. FISH tests are used particularly for ES/PNET tumors at unusual sites (extraskeletal ES/PNET).
| Extraskeletal ES/PNET | ▴Top |
Extraskeletal ES/PNETs are rare, aggressive and highly malignant soft tissue tumors indistinguishable morphologically from the skeletal types. Extraskeletal ES/PNET was firstly reported by Angervall and Enzinger in 1975 [10], and are characterized by high incidence of recurrence and distant metastasis. The most common sites to be involved by extraskeletal ES/PNET are lower extremities, retroperitoneal space, chest wall and gluteal region. Other less commonly involved organs include the kidneys, prostate, gastrointestinal (GI) tract, brain, adrenal glands, breast, ovaries and endometrium.
Primary gastric ES/PNET is an extremely rare entity, with a paucity of cases reported in the English literature.
| Primary Gastric ES/PNET | ▴Top |
Reviewing the current English literature reveals only eight cases of primary ES/PNET of the stomach [3, 4, 9, 11-15]. Of the eight patients, five were females while three were males (F/M ratio = 1.6:1), and the average age at presentation was 47 years old (range: 24 - 68). These findings contradict other extraskeletal subtypes of ES/PNET which usually affect younger patients with equal sex predilection. The most common clinical presentation was abdominal pain, noticed in six patients (75%), followed by vomiting and upper GI hemorrhage in three and two patients, respectively (Table 2 [3, 4, 9, 11-15]). Basma et al reported a case with an asymptomatic presentation, in which the tumor was incidentally found during upper endoscopy [10], while Kim et al reported a case of palpable abdominal mass on presentation [11]. Pre-operative diagnosis was not confirmed in most patients, as it was available in only three patients. The most common endoscopic finding for primary ES/PNET of the stomach was ulcerated protruding mass, detected in six patients (75%), with the antrum as the most common part to be affected (62%). Endoscopic findings were not available in one case in which the patient underwent surgical wedge resection of the tumor along with part of the lesser curvature with the pre-operative primary differential diagnosis being GI stromal tumor (GIST) [15]. Even though pre-operative diagnosis is not always available in all cases, the aforementioned case emphasizes the importance of pre-operative endoscopy during the workup. Primary gastric ES/PNETs of the lesser curvature and gastric body were diagnosed in one patient each. On abdominopelvic computed tomography (CT) scan, primary ES/PNET of the stomach usually presents as a large (9 - 11 cm) exophytic mass (most commonly) or as a circumferential hypodensic mass. All patients were treated by surgical resection, with the addition of neoadjuvant/ adjuvant chemotherapy in selected patients. Molecular study using FISH or reverse transcription polymerase chain reaction for EWSR1 gene rearrangement was done in six patients, and was found to be positive in five [3, 9, 12-14], and negative in one case [11]. Immunohistochemical staining for monoclonal antibody to CD 99 (HBA/71, 12E7 and 013) was positive in all reported cases, while it was found to be positive for vimentin and FL1 in four and three cases, respectively. These molecular studies indicate the importance of EWSR1 gene fusion translocation and monoclonal antibody for CD99 for precise diagnosis.
Click to view | Table 2. Reported Cases of Primary Ewing Sarcoma of the Stomach |
The extreme rarity of such entity, along with the absence of treatment guidelines, emphasizes the immense importance of a multidisciplinary team approach for optimal management. This multidisciplinary team should include GI surgeons, GI oncologists, gastroenterologists, pathologists and GI radiologists for optimal management.
| Conclusions | ▴Top |
Although primary ES/PNET of the stomach is an exceedingly rare entity, it should be part of the differential diagnosis of primary gastric tumors, especially in adult females with characteristic endoscopic ulcerated mass located at the gastric antrum. Multidisciplinary approach is advised for optimal care of such patients. Authors are encouraged to report such rare cases to gain knowledge about the different characteristics of these tumors.
Acknowledgments
None to declare.
Financial Disclosure
There was no specific funding source to be mentioned.
Conflict of Interst
The authors have no conflicts of interest to declare.
Informed Consent
Not applicable.
Author Contributions
Kenan Halloun and Subhi Mansour designed the research; Amir Farah, Meriam Obeid, Mira Damouny, Rozan Marjiyeh and Jawad Ghalia collected and analyzed data. Safi Khuri wrote and approved the final paper.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Ushigome S, Machinami R, Sorensen PH. Ewing sarcoma/primitive neuroectodermal tumour (PNET). In: Fletcher CDM, Unni KK, Mertens F, editors. World Health Organization Classification of Tumours. Pathology & Genetics of Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2002. p. 298-300.
- Durer S, Shaikh H. Ewing Sarcoma. [Updated 2020 Jun 23]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; Jan 2021.
- Soulard R, Claude V, Camparo P, Dufau JP, Saint-Blancard P, Gros P. Primitive neuroectodermal tumor of the stomach. Arch Pathol Lab Med. 2005;129(1):107-110.
doi pubmed - Hopp AC, Nguyen BD. Gastrointestinal: Multi-modality imaging of extraskeletal Ewing sarcoma of the stomach. J Gastroenterol Hepatol. 2019;34(8):1273.
doi pubmed - Applebaum MA, Worch J, Matthay KK, Goldsby R, Neuhaus J, West DC, Dubois SG. Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer. 2011;117(13):3027-3032.
doi pubmed - Salah S, Abuhijla FJ, Ismaeel T, et al. Outcomes of extra-skeletal versus skeletal Ewing sarcoma patients treated with standard chemotherapy protocol. J Clin Oncol. 2019;37:11027.
doi - Kempson RL, Fletcher CDM, Evans HL, et al. Tumors of the soft tissues. 3rd edition. Washington, DC: The Armed Forces Institute of Pathology; 2001. p. 444-452.
- Ambros IM, Ambros PF, Strehl S, Kovar H, Gadner H, Salzer-Kuntschik M. MIC2 is a specific marker for Ewing's sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewing's sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer. 1991;67(7):1886-1893.
doi - Khuri S, Gilshtein H, Sayidaa S, Bishara B, Kluger Y. Primary Ewing sarcoma/primitive neuroectodermal tumor of the stomach. Case Rep Oncol. 2016;9(3):666-671.
doi pubmed - Basma EE, Hajar H, Nabil M, Anis B, Samir B. Breast Ewing sarcoma/primitive neuroectodermal tumor: A case report and a review of the literature. Breast. 2012;3:WMC003792.
- Kim HS, Kim S, Min YD, Kee KH, Hong R. Ewing's sarcoma of the stomach; rare case of Ewing's sarcoma and suggestion of new treatment strategy. J Gastric Cancer. 2012;12(4):258-261.
doi pubmed - Rafailidis S, Ballas K, Psarras K, Pavlidis T, Symeonidis N, Marakis G, Sakadamis A. Primary Ewing sarcoma of the stomach—a newly described entity. Eur Surg Res. 2009;42(1):17-20.
doi pubmed - Song MJ, An S, Lee SS, Kim BS, Kim J. Primitive Neuroectodermal Tumor of the Stomach: A Case Report. Int J Surg Pathol. 2016;24(6):543-547.
doi pubmed - Maxwell AW, Wood S, Dupuy DE. Primary extraskeletal Ewing sarcoma of the stomach: a rare disease in an uncommon location. Clin Imaging. 2016;40(5):843-845.
doi pubmed - Kumar D, Kaur P, Khurana A, Chauhan AK, Sen R. Primary Ewing's sarcoma /primitive neuroectodermal tumor of stomach. Trop Gastroenterol. 2016;37(2):133-137.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.