

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 14, Number 3, June 2023, pages 178-187

Fluctuations in Gut Microbiome Composition During Immune Checkpoint Inhibitor Therapy
Joy Sarkara, l ![]() , Eduardo Cortes Gomezb, c, l
, Eduardo Cortes Gomezb, c, l ![]() , Takaaki Obad, e
, Takaaki Obad, e ![]() , Hongbin Chenf, g
, Hongbin Chenf, g ![]() , Grace K. Dyf, g
, Grace K. Dyf, g ![]() , Brahm H. Segalg, h, i
, Brahm H. Segalg, h, i ![]() , Marc S. Ernstofff, g, j
, Marc S. Ernstofff, g, j ![]() , Fumito Itoa, d, k, m
, Fumito Itoa, d, k, m ![]()
aDepartment of Surgical Oncology, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA
bDepartment of Biostatistics & Bioinformatics, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA
cDepartment of Biostatistics, School of Public Health and Health Professions, SUNY at Buffalo, NY, USA
dCenter for Immunotherapy, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA
eDivision of Breast and Endocrine Surgery, Department of Surgery, Shinshu University School of Medicine, Matsumoto, Japan
fDepartment of Medicine, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA
gDepartment of Medicine, University at Buffalo Jacobs School of Medicine and Biomedical Sciences, the State University of New York, Buffalo, NY, USA
hDepartment of Internal Medicine, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA
iDepartment of Immunology, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA
jNational Cancer Institute, Division of Cancer Treatment and Diagnosis, Bethesda, Maryland, USA
kDepartment of Surgery, Norris Comprehensive Cancer Center, Keck School of Medicine of USC, University of Southern California, Los Angeles, CA, USA
lThese authors contributed equally to the study.
mCorresponding Author: Fumito Ito, Department of Surgical Oncology, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA
Manuscript submitted March 20, 2023, accepted May 1, 2023, published online June 11, 2023
Short title: Gut Microbiota Dynamics During Immunotherapy
doi: https://doi.org/10.14740/wjon1587
| Abstract | ▴Top |
Background: Immune checkpoint inhibitors (ICIs) such as programmed cell death protein-1 (PD-1) inhibitors or PD-1 ligand-1 (PD-L1) inhibitors have led to remarkable improvement in outcomes of non-small cell lung cancer (NSCLC). Unfortunately, the significant benefits of ICI therapy are frequently limited by resistance to treatment and adverse effects, and the predictive value of pre-treatment tumor tissue PD-L1 expression is limited. Development of less invasive biomarkers that could identify responders and non-responders in early on-treatment could markedly improve the treatment regimen. Accumulating evidence suggests that baseline gut microbiota profile is associated with response to PD-1/PD-L1 blockade therapy. However, change in the gut microbiome composition during PD-1/PD-L1 blockade therapy and its relation to response remain unclear.
Methods: Here, we analyzed pre- and on-treatment fecal samples from five NSCLC patients receiving anti-PD-1 immunotherapy, alone or in tandem with chemotherapy, and performed 16S rRNA sequencing.
Results: The overall alpha diversity of the baseline gut microbiome was similar between three responders and two non-responders. While the gut microbiome composition remained stable overall during treatment (R2 = 0.145), responders showed significant changes in microbiome diversity between pre- and on-treatment samples during anti-PD-1 therapy compared to non-responders (P = 0.0274). Within the diverse microbiota, responders showed decreases in the abundance of genera Odoribacter, Gordonibacter, Candidatus Stoquefichus, Escherichia-Shigella, and Collinsella, and increase in abundance of Clostridium sensu stricto 1. In contrast, non-responders demonstrated on-treatment increases in genera Prevotella, Porphyromonas, Streptococcus, and Escherichia-Shigella, and decrease in abundance of Akkermansia.
Conclusions: This pilot study identified a substantial change in gut microbiome diversity between pre- and on-treatment samples in NSCLC patients responding to anti-PD-1 therapy compared to non-responders. Our findings highlight the potential utility of gut microbiota dynamics as a noninvasive biomarker to predict response to PD-1/PD-L1 blockade therapy for a wide variety of malignancies, which sets a path for future investigation in larger prospective studies.
Keywords: Gut microbiome; Biomarker; Immunotherapy; PD-1; Non-small cell lung cancer
| Introduction | ▴Top |
The paradigm of treatment of non-small cell lung cancer (NSCLC) has significantly evolved over the last several decades, in large part due to the development of immune checkpoint inhibitor (ICI) therapy [1, 2]. ICIs exert inhibitory signals on lymphocyte receptors or their ligands to unleash the anti-tumor immune response; among the most commonly used ICIs are monoclonal antibodies against programmed cell death protein 1 (PD-1), and its ligand PD-L1 [3, 4]. Whereas treatment of NSCLC with platinum-based chemotherapy only provided a modest improvement in survival compared to observation, treatment with PD-1/PD-L1 blockade therapy has demonstrated markedly improved prognosis [1, 5, 6]. However, many patients do not respond or experience progression of disease on immunotherapy due to resistance to treatment [1, 2]. Given that ICIs can be accompanied by serious immune-related adverse events, there is ongoing interest in identifying surrogate predictive markers which may play a role in a patient’s response to ICI treatment.
There are several biomarkers which correlate with response to anti-PD-1 therapy, such as PD-L1 expression in the tumor microenvironment (TME), CD8+ tumor infiltrating lymphocyte (TIL) density, and tumor mutational burden (TMB). However, predictive value of these markers on pre-treatment tumor tissue is limited because of dynamic change in the TME during ICI therapy [7-9]. Given that resistance to anti-PD-1 therapy is a dynamic process that evolves during the course of treatment [7-9], we sought to identify on-treatment biomarkers which can be monitored at several time points throughout treatment. While repeating tumor biopsies would provide the most accurate snapshot of the evolving TME [7-9], this is an invasive method which is impractical and potentially risk-prohibitive for visceral tumors.
In recent years, there has been a drastic growth of interest in how the gut microbiome affects outcomes of ICI therapy in various cancers [10-19]. The relationship between gut flora and the immune system is reciprocal in that microbiota shape host immunity by maintaining immune homeostasis, and the intestinal immune system functions on multiple levels to compartmentalize gut bacteria and minimize translocation [20]. The link between gut microbiota and ICI response has been highlighted by the fact that antibiotic use preceding ICI therapy is associated with worse outcomes [18, 19], and that fecal microbiota transplantation from ICI responders to non-responders can overcome primary resistance to therapy [21, 22]. Although these findings highlight the promise of strategies that target the gut microbiome as a predictive marker of response and the treatment of disease, it remains unclear whether the microbiome composition changes during treatment, and relationship between microbiota remodeling and response to ICI therapy remain unclear.
Improved understanding of the changes in gut microbiome in response to ICI therapy may provide new insights into the immune monitoring and mechanisms of anti-tumor effects with ICI therapy. Here, we conducted a pilot study where we assessed gut microbiota signatures in pre- and on-treatment stool samples from five NSCLC patients receiving anti-PD-1 therapy, alone or in combination with chemotherapy. Our results demonstrate that increased change of diversity of microbiota between pre- and on-treatment stool samples associates with response to anti-PD-1 therapy, and that serial fecal sampling may be a noninvasive method to predict response to ICI therapy.
| Materials and Methods | ▴Top |
Data reporting
Clinical samples were obtained from all participants enrolled during the study window. The study participants included a pool of both responders and non-responders. Due to the nature of this study, randomization could not be reasonably applied. The relevant biomarkers as detailed below were quantified at prespecified intervals during treatment as well as clinical response.
Study design, patients, and specimen collection
Patients at Roswell Park Comprehensive Cancer Center who received the anti-PD-1 antibody pembrolizumab, with or without chemotherapy (carboplatin and pemetrexed) for stages IIIA - IV NSCLC were eligible for inclusion in the study. We prospectively recruited patients for blood and stool sampling between February 2018 and October 2019. All participants gave informed consent for the collection and storage of blood and stool samples, and review of their medical records under the protocol was approved by the Institutional Review Board of Roswell Park Comprehensive Cancer Center. This study conformed to the Declaration of Helsinki. Prior to treatment, we obtained baseline samples of both peripheral blood (PB) and stool. PB was collected in ethylenediaminetetraacetic acid (EDTA)-containing tubes prior to each infusion, as well as every 3 weeks for 6 weeks. We collected a second stool sample within 3 weeks after the first cycle of the treatment. Ultimately, we accrued and included five NSCLC patients who received at least one dose of pembrolizumab, stool collection prior to and during treatment, and staging/surveillance imaging.
Flow cytometry
Using lymphocyte separation medium (Corning) density gradient centrifugation, we isolated peripheral blood mononuclear cells (PBMCs). Staining of CX3C chemokine receptor 1 (CX3CR1) in T cells was performed as described before [23, 24]. In brief, fresh or cryopreserved PBMCs were incubated with Fc block with human immunoglobulin G (IgG) (Sigma) at 12 mg/mL for 20 min. Anti-human CD3 (clone UCHT1), CD8 (clone RPA-T8), and CX3CR1 (clone 2A9-1) antibodies were obtained from BioLegend, and anti-CD4 (clone RPA-T4) antibody was from BD Biosciences for flow cytometry. LSRFortessa (BD) was used for sample acquisition, and FlowJo software v10.1.5 (FlowJo LLC) was used for sample analysis.
Assessment of response
After the initiation of either ICI alone or combination therapy with ICI and chemotherapy, clinical response was assessed during the first 12 weeks of treatment using immune response evaluation criteria in solid tumors (RECIST) (iRECIST) [25] criteria. Participants were denoted either responders or non-responders, with responders further subcategorized into complete response (CR) or partial response (PR), and non-responders subcategorized into stable disease (SD) or progressive disease (PD).
Immunohistochemical studies
The Dako Omnis platform (Agilent) with 22C3 pharmDx antibody was used to determine tumor expression of PD-L1, which was then scored by published guidelines [26].
Isolation of DNA from stool samples and 16S DNA sequencing
Qiagen DNeasy PowerSoil extraction kit following the manufacturer’s protocol was used to extract DNA from stool samples. We performed a two-step polymerase chain reaction (PCR) to build our sequencing libraries. A total of 25 ng of DNA were used in the first step (25 PCR cycles), and then amplify a target region (about 500 bp) of 16s V3 and V4 rDNA. Subsequently, the product from the first step was amplified with eight cycles of PCR using the Nextera Index kit (Illumina Inc.), which utilizes primers that aim for the overhang adaptor sequence incorporated during the first round of PCR. In the second PCR cycle, a unique combination of indexed tags was added to each sample, enabling library pooling and multiplex sequencing. Prior to the pooling step, each sample’s amplified DNA was visualized on a Tapestation 4200 D1000 tape (Agilent Technologies) to check the size, purity, and concentration of the expected product. Validated libraries were then pooled equal molar in a final resulting concentration of 4 nM in tris-HCl 10 mM, pH 8.5, before 2 × 300 cycle sequencing on a MiSeq (Illumina, Inc.) using the appropriate v3 reagents.
16S-seq taxonomic quantification
Paired-end fastq reads were demultiplexed, processed, and analyzed using QIIME v1.9.1 [27]. Then, operational taxonomic units (OTUs) are assigned using QIIME’s uclust-based [28] open-reference OTU-picking pipeline using SILVA [29] 16S rRNA reference (v132). Bacterial sequences were mapped using QIIME’s default alignment tool PyNAST [30]. Alignments were then refined by filtering out chimeric sequences using ChimeraSlayer. OTUs with less than 0.001% assigned were removed from individual samples, thus avoiding inflated alpha diversity estimates. Additional positive and negative control samples were examined against the whole batch and removed for downstream analyses.
Statistics
Gut microbiota were classified into OTUs at the genus level. Alpha-diversity scores were calculated using phyloseq package (v1.28.0) [31] to estimate the Observed, Chao1, Shannon and Simpson’s Reciprocal diversities. Mean alpha diversity scores estimates were obtained by performing 100 bootstrapped rarefactions to 130,000 sequences per sample. Group comparisons were performed using analysis of variance (ANOVA). Beta-diversity analysis was quantified with Bray-Curtis dissimilarity scores paired with MDS. Statistical group comparisons were performed using the permutational multivariate analysis of variance (PERMANOVA) procedure (running 5,000 permutations) implemented by the vegan package (v2.5.6) [32]. DESeq2 (v1.20.0) [33] was used to compare and detect differential abundance in OTUs between samples. Differential abundance results were visualized using dual-taxa plots and heatmaps reporting relevant OTUs selecting those having a P value < 0.05 and log2 fold-change > 2. Statistical analyses were performed in R (3.6.1).
| Results | ▴Top |
Patients, treatment outcomes, and biomarker performance
Baseline patient and treatment characteristics are summarized in Table 1. The five patients had a median age of 62, and four of five were female. The majority of patients had adenocarcinoma; one patient had NSCLC with giant cell features. One patient was in stage IIIA at diagnosis, and the remainder were stage IV. One patient received pembrolizumab alone, and the remainder of patients received a combination of carboplatin, pemetrexed, and pembrolizumab. PD-L1 expression tumor proportion score (TPS) was obtainable for four of the patients; one had a PD-L1 TPS ≥ 50%, and the remaining three patients had PD-L1 TPS between 1% and 49%. Two patients had a PR to treatment, one patient had a CR (Fig. 1a), and two patients had PD. The two patients who were deemed non-responders due to progression were both diagnosed with leptomeningeal disease on lumbar puncture; therefore, their imaging was non-contributory. Responders had median progression-free survival (PFS) and overall survival (OS) of 535 days while non-responders had median PFS of 108 days and OS of 129.5 days. Three responders had no gastrointestinal symptoms such as nausea, vomiting, constipation, and diarrhea within 12 weeks. Of two patients who did not respond to chemo-immunotherapy, one had mild nausea and constipation at 5 weeks, and the other developed immune-related gastritis causing nausea, emesis and constipation and was started on steroids at 9 weeks after beginning treatment. No patients had gastrointestinal symptoms at the time of on-treatment stool sample collection.
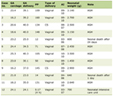 Click to view | Table 1. Demographic and Clinical Characteristics of Patients |
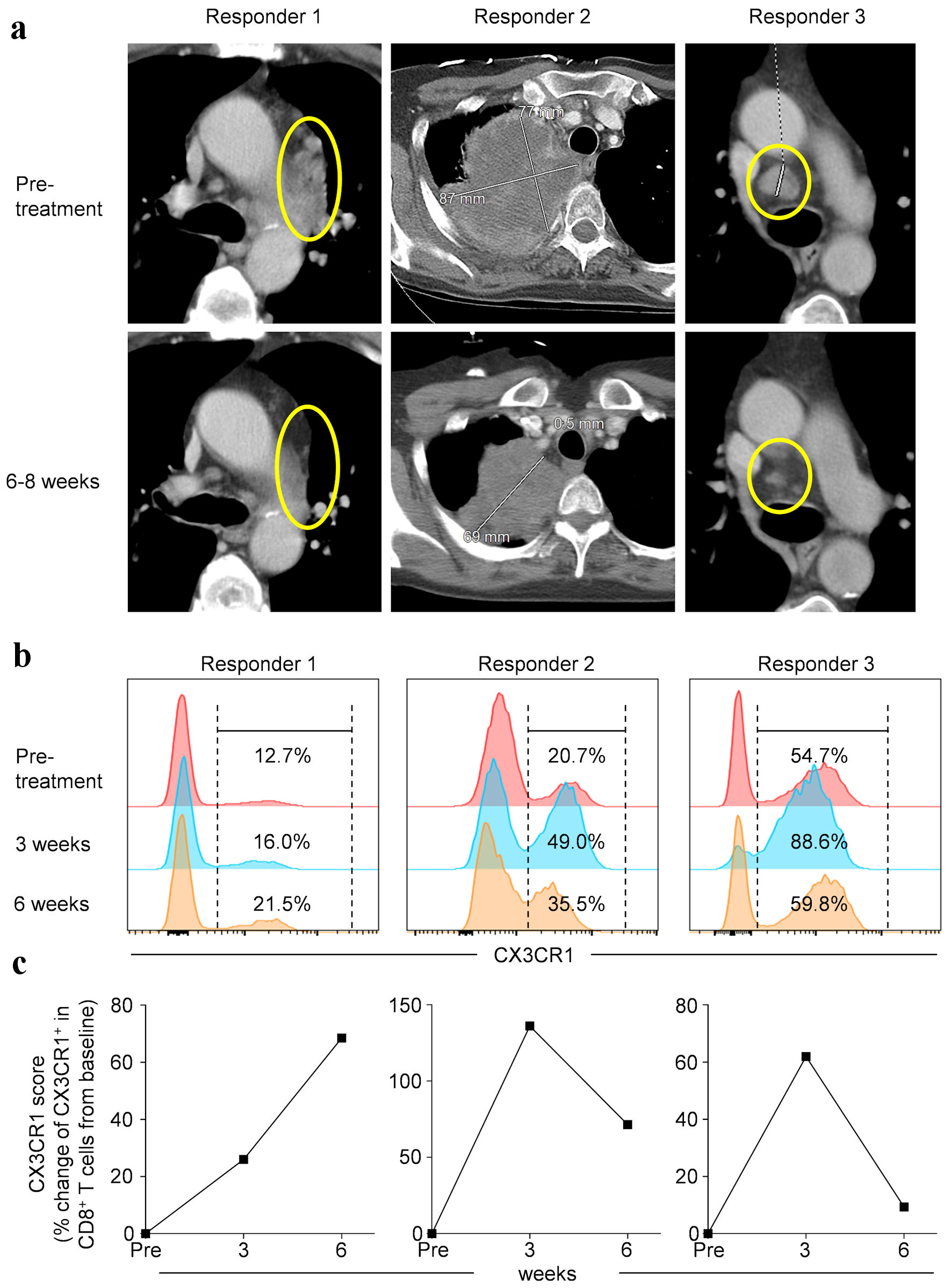 Click for large image | Figure 1. Pre- and on-treatment imaging study and circulating biomarker performance in non-small cell lung cancer (NSCLC) patients treated with anti-PD-1 therapy. (a) Contrast-enhanced cross sectional imaging obtained prior to and during treatment in three patients. Expression of CX3CR1 in peripheral blood CD8+ T cells (b) and % change of CX3CR1+ in CD8+ T cells from baseline (CX3CR1 score) (c) at different time points as indicated. PD-1: programmed cell death protein-1; CX3CR1: CX3C chemokine receptor 1. |
We have recently shown that expression of CX3CR1 on CD8+ T cells, marking T-cell differentiation [34], is linked to immunotherapy response in pre-clinical models and NSCLC patients treated with anti-PD-1/PD-L1 therapy [23]. In the study, a 20% rise in the CX3CR1 score (defined as the proportion of circulating CD8+ T cells expressing CX3CR1) from baseline predicted response to anti-PD-1/PD-L1 therapy as early as 3 weeks [23]. Therefore, in the current study we assessed the proportion of PB CD8+ T cells expressing CX3CR1 in the three patients who responded to treatment with pembrolizumab and calculated the CX3CR1 score in our patient cohort. We found that the PB CX3CR1+ CD8+ T cells substantially expanded at 3 weeks (Fig. 1b), resulting in at least 20% rise of the CX3CR1 score in all three responders (Fig. 1c). Although the CX3CR1 scores of the non-responders were not available for comparison, our findings indicate that response in these three patients is demonstrated not only by the imaging study (Fig. 1a) but also by circulating biomarker performance (Fig. 1b, c).
Fluctuations in gut microbiome in NSCLC patients during anti-PD-1 therapy
Next, we evaluated gut microbial signatures at baseline and early on-treatment in three responders and two non-responders. Across four different measures (Observed, Chao1, Simpson and Shannon) of alpha diversity, there were no baseline differences in the gut microbiomes of patients who did and did not respond to anti-PD-1 therapy (Fig. 2a).
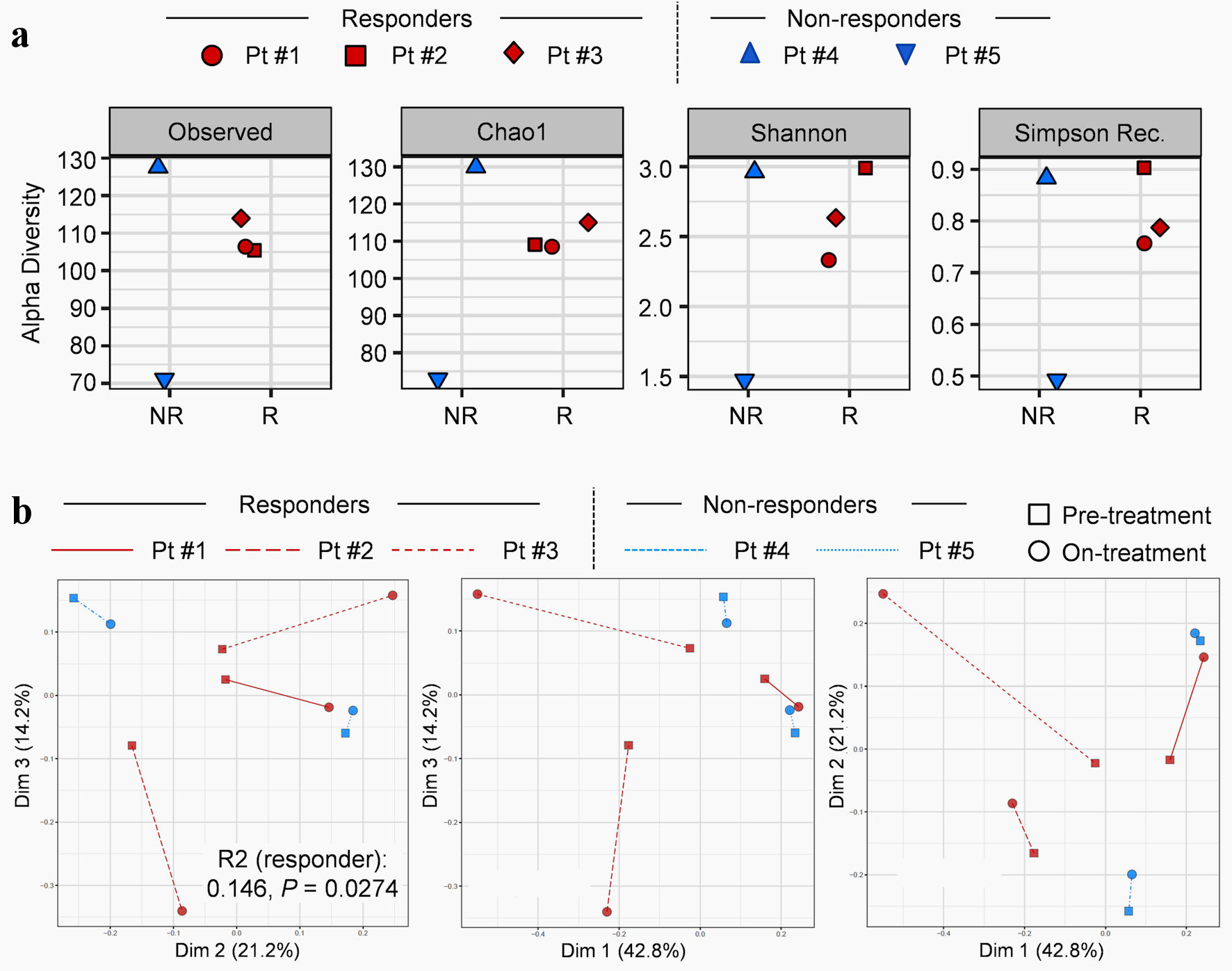 Click for large image | Figure 2. Fluctuations in gut microbiome in non-small cell lung cancer (NSCLC) patients during anti-PD-1 therapy. (a) Alpha diversities of baseline gut microbiome in responders (R) and non-responders (NR). First column: observed diversity reflects the total number of unique organisms. Second column: Chao1 diversity reflects total richness, weighted towards rare species. Third column: Shannon index reflects both richness and evenness of each sample. Fourth column: Simpson index reflects richness, weighted toward common species. (b) Beta-diversity using Bray-Curtis dissimilarity coupled with multidimensional scaling depicting pre- and post-treatment with anti-PD-1 immunotherapy. The first three pairwise principal components were displayed. P value was estimated from PERMANOVA using Bray-Curtis dissimilarity implemented by vegan R package (v2.5.6). Pt: patient; PD-1: programmed cell death protein-1; PERMANOVA permutational multivariate analysis of variance. |
The changes in gut microbiome beta diversity pre- and post-treatment were tested applying PERMANOVA using Bray-Curtis dissimilarity coupled with multidimensional scaling (MDS) (Fig. 2b). After accounting for treatment time points (P = 0.26) and subject effects (R2: 0.547, P = 0.0008), responders demonstrated a significant change in diversity of gut microbial between pre- and on-treatment samples compared to non-responders (R2: 0.146, P = 0.0274).
Differential on-treatment change in microbial profiles between responders and non-responders
We next assessed microbial composition in responders (Fig. 3a) and non-responders (Fig. 3b). Overall change in genus composition for each subject is depicted in Figure 3c. Responders demonstrated statistically significant post-treatment decreases in the abundance of genera Odoribacter, Gordonibacter, Candidatus stoquefichus, Escherichia-Shigella, and Collinsella, and increase in abundance of Clostridium sensu stricto 1 (Fig. 3a, c). In contrast, non-responders demonstrated statistically significant post-treatment increases in genera Prevotella, Porphyromonas, Streptococcus, and Escherichia-Shigella, and decrease in abundance of Akkermansia (P < 0.05) (Fig. 3b, c). These findings suggest that on-treatment change in microbial profiles was different between responders and non-responders.
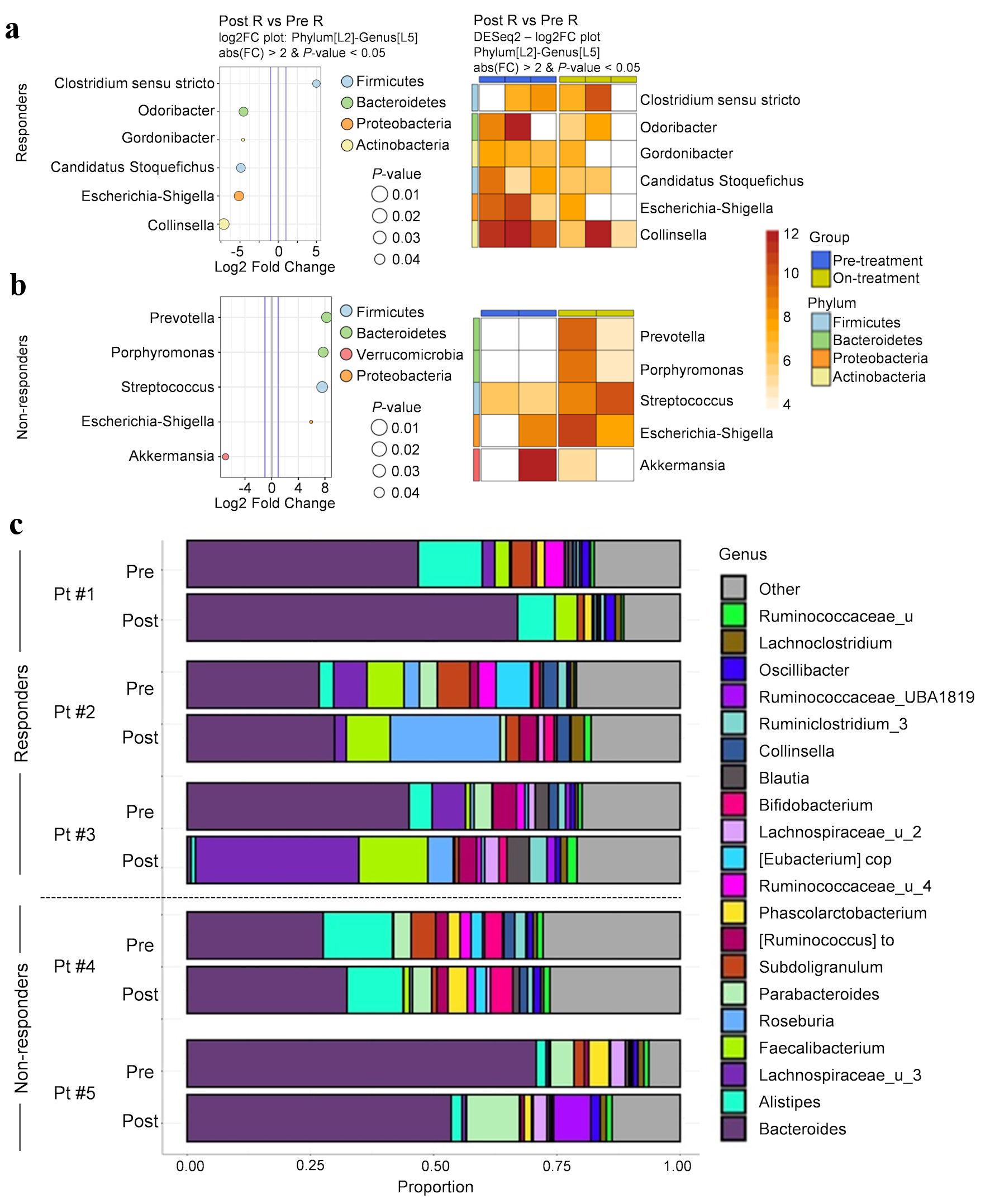 Click for large image | Figure 3. Substantial change in microbial profiles in NSCLC patients responding to anti-PD-1 therapy. (a, b) Significantly abundant genera found in pre- and on-treatment microbial composition between responders (a) and non-responders (b). Heatmap demonstrating significant differences in pre- and post-treatment microbial composition for each subject (n = 5); each pair of blue and yellow columns represents one subject. A variance-stabilization transformation (implemented by DESeq2) was used for the taxa abundance values. Darker shades represent higher differential abundance. (c) Genus composition pre- and on-treatment for all subjects. Pt: patient; NSCLC: non-small cell lung cancer; PD-1: programmed cell death protein-1. |
| Discussion | ▴Top |
Cancer immunotherapies that reinvigorate anti-tumor T cells by blocking the PD-1/PD-L1 axis have shown promise in improving outcomes for many cancers. Despite this, response to immunotherapy is far from uniform, and immune-related adverse effects are not negligible. Predicting and identifying which patients undergoing immunotherapy will response to treatment remains a major challenge. While intratumoral PD-L1 expression has been used for the selection of NSCLC patients, PD-L1-negative tumors may respond to anti-PD-1 therapy, and some tumors with high PD-L1 expression still demonstrate primary resistance [35, 36]. Of late, evidence is mounting which substantiates the association between the gut microbiome and response to ICI therapy [10-19]. The gut microbiome and microbiome-derived metabolites could impact immune cell recruitment within both the TME as well as systemically, which in turn modulates the efficacy and adverse events associated with ICI therapy. As an example, use of antibiotics has been shown to reduce the efficacy of ICIs in a number of cancers [37, 38]. However, specific gut bacterial metabolites such as short-chain fatty acids can augment regulatory T-cell responses that protect against autoimmunity but can also impair antitumor immunity [39]. Furthermore, evolution of gut microbiome during ICI therapy and its relation to response are incompletely understood. Here, we describe differential change in microbiota signatures during anti-PD-1 therapy between responders and non-responders. These findings imply that increased change in gut microbiome diversity might correlate with response to anti-PD-1 therapy in NSCLC patients.
Correlation between alpha diversity of species within the gut microbiome and the responses to ICI therapy remains unclear. We found no significant difference in the alpha diversity between responders and non-responders in our cohort, which is consistent with previous studies [17, 18, 40, 41]. However, other studies have shown that increased alpha diversity in a favorable group of patients treated with PD-1/PD-L1 blockade therapy [11, 19, 42]. These inconsistent results might be associated with limited sample size as well as the type of ICIs and/or cancer. Additional work is required to determine the role of alpha diversity at baseline in response and resistance to ICI therapy.
Prior studies have examined the correlation between specific gut bacterial species and response to ICI therapy [11, 43]. The abundance of Bifidobacterium, Coriobacteria, Ruminococcae, Prevotellaceae, and Lachnospiraceae have been linked to good response to ICI therapy [22, 41], while non-responders have been found to have gut microbiome abundance of species such as Bacteroides and Escherichia coli [44]. Specifically, in recent years, numerous studies have demonstrated that abundance of Akkermansia is associated with favorable outcomes with ICI therapy [10, 12, 41]. In this study, we found that non-responders demonstrated substantial on-treatment increases in genera Prevotella, Porphyromonas, Streptococcus, and Escherichia-Shigella, and a decrease in abundance of Akkermansia during treatment. The increase in Escherichia abundance and decrease in Akkermansia abundance in correlation with poorer clinical outcome is in line with previously published findings [10, 12, 41]. However, more work is needed to conclusively define the association between the presence of specific bacteria at baseline or on-treatment and response to ICI therapy.
While multiple studies have evaluated the baseline gut microbiome composition as a biomarker for predicting anti-PD-1 efficacy in NSCLC, few have examined the change in microbial composition during treatment as a predictor of response to treatment. Andrews et al demonstrated that mice injected with combined immune checkpoint blockade developed a relative abundance of Bacteroides intestinalis compared to baseline [40]. As part of a larger study of melanoma patients undergoing anti-PD-1 immunotherapy, Gopalakrishnan et al showed that in three patients who provided fecal samples before and during treatment, their gut microbial diversity and composition remained stable over time [11]; however, it was not reported whether these patients were responders or non-responders. In clinical studies, Zheng et al reported in a study of eight patients with hepatocellular carcinoma (HCC) treated with anti-PD-1 antibody camrelizumab that responders demonstrated a relatively stable gut microbial composition, while non-responders showed more dramatic shifts in composition including increase of Proteobacteria [45]. The findings from this latter study contrast with those in our present work, as we found that responders demonstrated a larger overall fluctuation in gut microbiome composition compared to non-responders. The reason for this discrepancy is unclear, but these conflicting observations are likely a result of differences in the type of cancer and limited sample size. Nonetheless, in the numerous studies which have evaluated the link between specific gut microbes with response to ICI therapy, countless bacterial species have been associated with immunotherapy response or lack thereof, suggesting that no single, or even few, species can be considered a reliable biomarker of predicting ICI response. Rather, the combination of these studies implies that the relationship between gut microbiome composition and response to immunotherapy is more complex. Our findings suggest that the degree of change in overall gut microbiome diversity and composition during anti-PD-1 therapy may be a useful biomarker in and of itself, in comparison to baseline gut microbial profiles.
There are several limitations to this study. The main limitation was the small sample size due to the nature of pilot study. Collecting longitudinal stool samples was logistically difficult, and only one on-treatment stool sample was available for each patient to analyze in our cohort. Inherent to pilot studies, our sample size was insufficient to draw definitive conclusions on the correlation between change in the gut microbiome composition and the response to immunotherapy. Moreover, we had no data on change in the frequency of PB CX3CR1+ CD8+ T cells for the two non-responders, which limited our ability to draw a correlation between gut microbiome fluctuations and PB biomarkers. The CX3CR1 score was the only circulating biomarker that we evaluated, and additional work with multicolor flowcytometric analysis of PBMC and/or multi-omics approach of serum/plasma samples are needed to elucidate relationship between evolution of systemic immune response and gut microbiota signatures. Chemotherapy could induce gastrointestinal toxicity and/or shape intestinal microbiota [46]. Although none of the patients in the present study developed gastrointestinal symptoms at the time of on-treatment stool collection, the impact of the chemotherapy component of the treatment regimen on gut microbiome remains unclear. Nevertheless, this pilot study delivers potential utility of combined analysis of pre- and on-treatment gut microbiome and provides rationale for a longitudinal stool sampling during cancer immunotherapy.
In conclusion, we found in a study of five patients with NSCLC undergoing treatment with anti-PD-1 therapy that response to ICI treatment correlated with a larger change in gut microbial diversity compared to non-responders. This is one of very few studies to examine the longitudinal fluctuation in gut microbial diversity during immunotherapy for NSCLC, which highlights a potential focus for future larger-scale studies.
Acknowledgments
We are very grateful to all patients who generously contributed samples and participated in the study. We thank Dr. Prashant Singh (Roswell Park) for technical assistance.
Financial Disclosure
This work was supported by National Cancer Institute (NCI) grants P30CA016056 involving the use of Roswell Park’s Flow and Image Cytometry, Biostatistics & Statistical Genomics Shared Resource, and Data Bank and Biorepository Shared Resource. This work was supported by Roswell Park Alliance Foundation, Department of Defense Lung Cancer Research Program (LC180245), NCI grant, K08CA197966, R01CA255240-01A1 (F. Ito), R01CA188900 and R01CA267690 (B.H. Segal), and Uehara Memorial Foundation (T. Oba).
Conflict of Interest
The authors declare no potential conflicts of interest.
Informed Consent
Informed consent was obtained.
Author Contributions
JS analyzed data and wrote the manuscript. ECG analyzed data and reviewed the manuscript. TO performed experiments, acquired flow cytometry data, and revised the article. HC and GKD recruited patients and reviewed the manuscript. BHS and MSE assisted in acquisition of patient samples and revised the manuscript. FI developed the concept, managed the project, analyzed data, revised the manuscript, coordinated author activities, and provided final approval of the version to be submitted.
Data Availability
Raw and processed 16S rRNA sequencing data supporting the findings of this study are available via BioProject PRJNA919051. Other Source data are provided with this paper. All data generated and analyzed are available from the corresponding author upon reasonable request.
Abbreviations
ANOVA: analysis of variance; CD: cluster of differentiation; CR: complete response; CX3CR1: CX3C chemokine receptor 1; DNA: deoxyribonucleic acid; EDTA: ethylenediaminetetraacetic acid; ICI: immune checkpoint inhibitor; IgG: immunoglobulin G; MDS: multidimensional scaling; NSCLC: non-small cell lung cancer; OS: overall survival; OTU: operational taxonomic unit; PB: peripheral blood; PBMC: peripheral blood mononuclear cell; PCR: polymerase chain reaction; PD: progressive disease; PD-1: Programmed cell death protein-1; PD-L1: PD-1 ligand-1; PERMANOVA: permutational multivariate analysis of variance; PFS: progression-free survival; PR: partial response; RECIST: response evaluation criteria in solid tumors; RNA: ribonucleic acid; SD: stable disease; TMB: tumor mutational burden; TME: tumor microenvironment; TIL: tumor infiltrating lymphocyte; TPS: tumor proportion score
| References | ▴Top |
- Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, Gottfried M, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823-1833.
doi pubmed - Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, Molina J, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387(10027):1540-1550.
doi pubmed - Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455-2465.
doi pubmed pmc - Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443-2454.
doi pubmed pmc - Felip E, Altorki N, Zhou C, Csoszi T, Vynnychenko I, Goloborodko O, Luft A, et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB-IIIA non-small-cell lung cancer (IMpower010): a randomised, multicentre, open-label, phase 3 trial. Lancet. 2021;398(10308):1344-1357.
doi pubmed - Forde PM, Spicer J, Lu S, Provencio M, Mitsudomi T, Awad MM, Felip E, et al. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N Engl J Med. 2022;386(21):1973-1985.
doi pubmed pmc - Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171(4):934-949.e916.
doi pubmed pmc - Vilain RE, Menzies AM, Wilmott JS, Kakavand H, Madore J, Guminski A, Liniker E, et al. Dynamic changes in PD-L1 expression and immune infiltrates early during treatment predict response to PD-1 blockade in melanoma. Clin Cancer Res. 2017;23(17):5024-5033.
doi pubmed - Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016;6(8):827-837.
doi pubmed pmc - Derosa L, Routy B, Thomas AM, Iebba V, Zalcman G, Friard S, Mazieres J, et al. Intestinal Akkermansia muciniphila predicts clinical response to PD-1 blockade in patients with advanced non-small-cell lung cancer. Nat Med. 2022;28(2):315-324.
doi pubmed pmc - Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97-103.
doi pubmed pmc - Grenda A, Iwan E, Chmielewska I, Krawczyk P, Giza A, Bomba A, Frak M, et al. Presence of Akkermansiaceae in gut microbiome and immunotherapy effectiveness in patients with advanced non-small cell lung cancer. AMB Express. 2022;12(1):86.
doi pubmed pmc - Lee PC, Wu CJ, Hung YW, Lee CJ, Chi CT, Lee IC, Yu-Lun K, et al. Gut microbiota and metabolites associate with outcomes of immune checkpoint inhibitor-treated unresectable hepatocellular carcinoma. J Immunother Cancer. 2022;10(6):e004779.
doi pubmed pmc - Martini G, Ciardiello D, Dallio M, Famiglietti V, Esposito L, Corte CMD, Napolitano S, et al. Gut microbiota correlates with antitumor activity in patients with mCRC and NSCLC treated with cetuximab plus avelumab. Int J Cancer. 2022;151(3):473-480.
doi pubmed pmc - Thompson NA, Stewart GD, Welsh SJ, Doherty GJ, Robinson MJ, Neville BA, Vervier K, et al. The MITRE trial protocol: a study to evaluate the microbiome as a biomarker of efficacy and toxicity in cancer patients receiving immune checkpoint inhibitor therapy. BMC Cancer. 2022;22(1):99.
doi pubmed pmc - Yin H, Yang L, Peng G, Yang K, Mi Y, Hu X, Hao X, et al. The commensal consortium of the gut microbiome is associated with favorable responses to anti-programmed death protein 1 (PD-1) therapy in thoracic neoplasms. Cancer Biol Med. 2021;18(4):1040-1052.
doi pubmed pmc - Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, Luke JJ, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104-108.
doi pubmed pmc - Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, Fluckiger A, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359(6371):91-97.
doi pubmed - Hakozaki T, Richard C, Elkrief A, Hosomi Y, Benlaifaoui M, Mimpen I, Terrisse S, et al. The gut microbiome associates with immune checkpoint inhibition outcomes in patients with advanced non-small cell lung cancer. Cancer Immunol Res. 2020;8(10):1243-1250.
doi pubmed - Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336(6086):1268-1273.
doi pubmed pmc - Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, Adler K, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. 2021;371(6529):602-609.
doi pubmed - Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, Deblasio RN, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. 2021;371(6529):595-602.
doi pubmed pmc - Yamauchi T, Hoki T, Oba T, Jain V, Chen H, Attwood K, Battaglia S, et al. T-cell CX3CR1 expression as a dynamic blood-based biomarker of response to immune checkpoint inhibitors. Nat Commun. 2021;12(1):1402.
doi pubmed pmc - Yamauchi T, Hoki T, Oba T, Saito H, Attwood K, Sabel MS, Chang AE, et al. CX3CR1-CD8+ T cells are critical in antitumor efficacy but functionally suppressed in the tumor microenvironment. JCI Insight. 2020;5(8):e133920.
doi pubmed pmc - Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar S, Lin NU, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017;18(3):e143-e152.
doi pubmed pmc - Dako. PD-L1 IHC 28-8 pharmDx: non-squamous non-small cell lung cancer [interpretation manual]. Santa Clara, CA: Dako. 2017.
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335-336.
doi pubmed pmc - Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460-2461.
doi pubmed - Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(Database issue):D590-D596.
doi pubmed pmc - Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26(2):266-267.
doi pubmed pmc - McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217.
doi pubmed pmc - Oksanen J, Kindt R, Legendre P, O’Hara B, Stevens MHH, Oksanen MJ, et al. The vegan package. Community ecology package. 2007;10(631-637):719.
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
doi pubmed pmc - Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, Garg R, et al. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity. 2016;45(6):1270-1284.
doi pubmed pmc - Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol. 2021;16:223-249.
doi pubmed - Vesely MD, Zhang T, Chen L. Resistance mechanisms to anti-PD cancer immunotherapy. Annu Rev Immunol. 2022;40:45-74.
doi pubmed - Derosa L, Hellmann MD, Spaziano M, Halpenny D, Fidelle M, Rizvi H, Long N, et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann Oncol. 2018;29(6):1437-1444.
doi pubmed pmc - Pinato DJ, Gramenitskaya D, Altmann DM, Boyton RJ, Mullish BH, Marchesi JR, Bower M. Antibiotic therapy and outcome from immune-checkpoint inhibitors. J Immunother Cancer. 2019;7(1):287.
doi pubmed pmc - Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504(7480):451-455.
doi pubmed pmc - Andrews MC, Duong CPM, Gopalakrishnan V, Iebba V, Chen WS, Derosa L, Khan MAW, et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat Med. 2021;27(8):1432-1441.
doi pubmed - Peng Z, Cheng S, Kou Y, Wang Z, Jin R, Hu H, Zhang X, et al. The gut microbiome is associated with clinical response to anti-PD-1/PD-L1 immunotherapy in gastrointestinal cancer. Cancer Immunol Res. 2020;8(10):1251-1261.
doi pubmed - Jin Y, Dong H, Xia L, Yang Y, Zhu Y, Shen Y, Zheng H, et al. The diversity of gut microbiome is associated with favorable responses to anti-programmed death 1 immunotherapy in Chinese patients with NSCLC. J Thorac Oncol. 2019;14(8):1378-1389.
doi pubmed - Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, Boselli L, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2017;28(6):1368-1379.
doi pubmed - Sevcikova A, Izoldova N, Stevurkova V, Kasperova B, Chovanec M, Ciernikova S, Mego M. The impact of the microbiome on resistance to cancer treatment with chemotherapeutic agents and immunotherapy. Int J Mol Sci. 2022;23(1):488.
doi pubmed pmc - Zheng Y, Wang T, Tu X, Huang Y, Zhang H, Tan D, Jiang W, et al. Gut microbiome affects the response to anti-PD-1 immunotherapy in patients with hepatocellular carcinoma. J Immunother Cancer. 2019;7(1):193.
doi pubmed pmc - Oh B, Boyle F, Pavlakis N, Clarke S, Guminski A, Eade T, Lamoury G, et al. Emerging evidence of the gut microbiome in chemotherapy: a clinical review. Front Oncol. 2021;11:706331.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.