

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 14, Number 6, December 2023, pages 464-475

Circadian Clock REV-ERBs Agonist SR9009 Induces Synergistic Antitumor Activity in Multiple Myeloma by Suppressing Glucose-Regulated Protein 78-Dependent Autophagy and Lipogenesis
Rui Wanga, b, e, Shu Ling Liuc, e, Quan Quan Guoa, b, Xiao Hong Shid, Mei Mei Mad, f
aDepartment of Hematology, The Second Affiliated Hospital of Soochow University, No. 1055, San Xiang Road, Gu Su District, Su Zhou City, China
bDepartment of Oncology, Suqian Affiliated Hospital of Xuzhou Medical University, No. 138, Huanghe Road, Su Cheng District, Suqian City, China
cDepartment of Radiotherapy, Nanjing Jinling Hospital, Affiliated Hospital of Medical School, Nanjing University, No.305, Zhong Shan Road, Xuan wu District, Nanjing City, China
dDepartment of Pathology, Suqian Affiliated Hospital of Xuzhou Medical University, No. 138, Huanghe Road, Su Cheng District, Suqian City, China
eThese authors contributed equally to this article.
fCorresponding Author: Mei Mei Ma, Department of Pathology, Suqian Affiliated Hospital of Xuzhou Medical University, No. 138, Huanghe Road, Su Cheng District, Suqian City, China. E-mail:
Manuscript submitted July 21, 2023, accepted September 26, 2023, published online October 21, 2023
Short title: SR9009 Induces Synergistic Antitumor Activity in MM
doi: https://doi.org/10.14740/wjon1681
| Abstract | ▴Top |
Background: Proteasome inhibitors, such as bortezomib, have demonstrated efficacy in the therapeutic management of multiple myeloma (MM). However, it is important to note that these inhibitors also elicit endoplasmic reticulum stress, which subsequently triggers the unfolded protein response (UPR) and autophagy, which have been shown to facilitate the survival of tumor cells. The disruption of the circadian clock is considered a characteristic feature of cancer. However, how disrupted circadian clock intertwines with tumor metabolism and drug resistance is not clearly clarified. This work explores the antitumor effectiveness of bortezomib and the circadian clock agonist SR9009, elucidating their impact on glucose-regulated protein 78 (GRP78), the autophagy process, and lipogenesis.
Methods: The antitumor effects of bortezomib and SR9009 were evaluated using human MM cell lines (RPMI8226 and U266) in vitro and in vivo nonobese diabetic/severe combined immunodeficient (NOD/SCID) murine xenograft MM model. The assessment of cell viability was conducted using the cell counting kit-8 (CCK8) method, whereas the measurement of cell proliferation was performed with the inclusion of EdU (5-ethynyl-2’-deoxyuridine). Apoptosis was assessed by flow cytometry. The cells were transduced using adenovirus-tf-LC3, which was labeled with dual fluorescence. Subsequently, confocal imaging was employed to observe and examine the autophagosomes. REV-ERBα knockdown leads to upregulation of ATG5 and BENC1 at the protein level with immunoblot. Changes in the expression levels of GRP78, LC3, stearoyl-CoA desaturase 1 (SCD1), and fatty acid synthase (FASN) were assessed through the utilization of quantitative real-time polymerase chain reaction (qRT-PCR) and western blotting.
Results: Our results showed that both bortezomib and circadian clock REV-ERBs agonist SR9009 decreased MM viability, proliferation rate and induced an apoptotic response in a dose-dependent manner in vitro. However, the two differ greatly in their mechanisms of action. Bortezomib upregulated GRP78 and autophagy LC3, while circadian clock agonist SR9009 inhibited GRP78 and autophagy LC3. Combined SR9009 with bortezomib induced synergistic cytotoxicity against MM cells. REV-ERBα knockdown lead to upregulation of ATG5, BENC1 and significant upregulation of FASN, and SCD1. Mechanically, SR9009 inhibited the core autophagy gene ATG5 and BECN1, and two essential enzymes for de novo lipogenesis FASN and SCD1. SR9009 had synergistic effect with bortezomib and slowed down murine xenograft models of human MM tumor growth in vivo.
Conclusions: Taken together, these results demonstrated that the circadian clock component REV-ERBs agonist SR9009 could inhibit GRP78-induced autophagy and de novo lipogenesis processes and had a synergistic effect with proteasome inhibitors in both in vitro and in vivo models of MM. Our findings shed light on how a disrupted circadian clock interacts with metabolic mechanisms to shape proteasome inhibitor drug resistance and suggest that SR9009 may be able to overcome the inherent drug resistance of proteasome inhibitors.
Keywords: Autophagy; Circadian clock; Lipogenesis; Multiple myeloma; SR9009
| Introduction | ▴Top |
In the clinic, proteasome inhibitors have significantly enhanced the overall survival rates of multiple myeloma (MM) patients [1, 2]. Proteasome inhibitors are unquestionably effective in the treatment of MM, but they also induce endoplasmic reticulum (ER) stress, resulting in the unfolded proteins response (UPR) and autophagy, which are cell survival coping mechanisms [3]. Glucose-regulated protein 78 (GRP78), also known as BiP, is a central regulator of ER function because of its roles in protein folding and assembly, targeting misfolded protein for degradation, ER Ca2+-binding, and regulating the activation of transmembrane ER stress sensors. ER stress associated with GRP78, and tumor cell autophagy are the primary causes of drug resistance, treatment failure, and tumor relapse [4-6]. It has been discovered that tumor intrinsic autophagy promotes immune evasion via the engulfment and digestion of tumor debris and major histocompatibility complex (MHC)1 antigens by LC3 proteins [7, 8], which helps to explain why a majority of patients with MM receiving therapy with proteasome inhibitors inevitably relapse after variable remission periods [9].
Due to its role in regulating ER stress sensing and protein folding, GRP78 is considered a crucial regulator of ER function. Proteasome inhibitors activate the unfolded protein response (UPR) to promote the survival of tumor cells [10, 11]. GRP78 facilitates cytoprotective autophagy via the maintenance of ER structural integrity and is required for UPR activation [12]. Overexpression of the HSPA5 gene, which encodes GRP78, is associated with a poor prognosis [13-15] and enhances proliferation of several tumor cells. HSPA5 suppression inhibits the growth of metastatic tumors in xenograft models [16]; GRP78 activation of the PI3K/Akt pathway supports cancer cell growth [17]. In newly diagnosed myeloma patient tumors, gene expression analysis revealed a correlation between overexpression of the HSPA5 gene encoding GRP78 and a diminished clinical response to treatment with bortezomib [3]. Previous research demonstrated that cells transfected with shRNA to reduce HSPA5 did not form autophagosomes and displayed decreased viability following bortezomib therapy compared to control transfected cells [3], suggesting that GRP78-ER stress is needed for autophagosome formation. Furthermore, previous research indicates that HSPA5 overexpression is associated with bortezomib tolerance in individuals who do not respond to treatment [3, 6].
Proliferation of cells and metabolism are governed by the daily cycles of the circadian clock [18]. These alterations are hallmarks of cancer [19]. Cancer cells are characterized by a disruption of the circadian clock. REV-ERBs consist of the circadian clock elements REV-ERBα (also known as NR1D1) and REV-ERBβ (also known as NR1D2) [20, 21]. Reduced REV-ERBs failed to inhibit the downstream BMAL1 and CLOCK, which frequently results in uncontrolled tumor growth and apoptosis resistance [22]. This raises the possibility that pharmacological modulation of the circadian machinery may be an effective cancer treatment, including MM.
| Materials and Methods | ▴Top |
Myeloma cell lines
The human MM cell lines U266 and RPIM8226 were purchased from the Chinese Academy of Sciences (Shanghai, China). Two cell types were cultured in RPMI with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin (Hyclone, USA) at 37 °C in 5% CO2. All cell lines were routinely tested with the MycoAlert Mycoplasma Detection Kit (Lonza) for the presence of mycoplasma.
Chemicals and reagents
Proteasome inhibitor was purchased from ActiveBiochem (Maplewood, NJ, USA), SR9009 was from Sigma-Aldrich (MCE: HY-16989), GRP78 antibodies from Affinity (AF5366), human specific antibodies to LC3 were from Proteintech (14600-1-AP), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from Proteintech (60004-1-Ig).
Cell counting kit-8 (CCK8) for cell viability assay
Cells were seeded into 96-well plates with the indicated concentrations of bortezomib, 15 µL of CCK8 solution was added into each well, and the plate was incubated at 37 °C with 5% CO2 for 4 h. Cell viability (mean ± standard deviation (SD)) was determined using absorbance from untreated cells and was measured at 450 nm in a spectrophotometric micro plate reader (Thermo MK3).
Flow cytometry apoptosis
The stained cells were evaluated using an LSR-II flow cytometer (BD Biosciences, San Jose, CA, USA): they had been treated in 5 µL of annexin V-fluorescein isothiocyanate (V-FITC) (Bestbio 401006) for 15 min and 10 µL of propidium iodide (PI) for another 10 min at room temperature in darkness.
NR1D1 knockdown
Lipofectamine RNAiMAX by Thermo Fisher Scientific was utilized to transfect cells with siRNA oligonucleotides purchased from Gima. U266 cells were transiently transfected with siNR1D1 plasmid.
Immunoblots
Add pre-chilled radioimmunoprecipitation assay (RIPA) lysis buffer containing phenylmethanesulfonyl fluoride (PMSF) to the obtained cell samples, mix thoroughly, centrifuge at 12,000g for 10 min after complete lysis, collect the supernatant, quantify protein with a bicinchoninic acid (BCA) reagent, and heat the protein to denature it. Prepare polyacrylamide gel and electrophoresis, then transfer the membrane. After separately incubating the membrane with GRP78, LC3, REV-ERBα, REV-ERβ, and a second antibody, enhanced chemiluminescence (ECL) reagent was added to the membrane for imaging, and images were captured using an integrated chemiluminescence instrument. In the “Chemicals and reagents” section, the antibodies utilized were described.
Immunostaining and confocal microscopy
To perform caspase 3, TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling), and GRP78 immunostaining, 100 µL of cell suspension from each of the groups was applied onto a glass slide after MM cells were incubated with bortezomib or SR9009 for 24 h. Cells were permeabilized with 0.2% triton X100 and fixated for 30 min with 4% paraformaldehyde. A primary antibody was used to stain the cells overnight, followed by a fluorescent secondary antibody. Hoechst was employed to identify the nuclei. Immunostaining was performed using cleaved caspase 3 (Cell Signaling No. 9664 1:200) and TUNEL (Cell Signaling No. 9635 1:200) antibodies to analyze apoptosis.
Detection of autophagy flux
Adenovirus which has dual-fluorescence labeling of mRFP-GFP-LC3 fusion protein was designed and purchased from Hanbio Technology (Shanghai, China). Autophagy double-labeled adenovirus was used to infect U266 cells at a multiplicity of infection (MOI) of 100. After 48 h of virus infection, each experimental group’s samples were administered in separate plates. After 24 h of incubation with SR9009, bortezomib, or both, the cells were observed using confocal microscopy to detect autophagy. To count LC3B puncta, the “analyze particles” function in ImageJ was used to determine a threshold that minimized the effect of background signal on the projected stack. The 3D Objects Counter was used to analyze the intensity of fluorescence.
Quantitative real-time polymerase chain reaction (qRT-PCR)
qRT-PCR comprises primarily of four steps: total RNA extraction, cDNA synthesis, PCR, and real-time PCR data processing. According to the manufacturer’s instructions, total RNA was extracted from cells using RNAeasy (Qiagen) and treated with DNase for reverse transcription. qScript cDNA Super Mix (Quanta Biosciences) was utilized to generate cDNA, which was then utilized as a template in RT-PCR reactions with specific primers on an ABI 7500HT Fast Real-Time PCR System. The data were analyzed with the software Bio-Rad CFX Manager. In the same sample, the mRNA level was normalized against GAPDH.
The primers utilized in reverse transcription PCR were designed using Primer Premier 5.0 software and synthesized by Shanghai Shenggong Company. Human primer sequences for qRT-PCR are shown in Table 1.
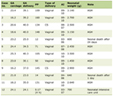 Click to view | Table 1. Human Primer Sequences for qRT-PCR |
Adenosine triphosphate (ATP) production assay
ATP production was measured with an ATP Assay Kit (Solarbio, BC0300) in which 25 µg of mitochondrial protein was incubated with ATP assay mix.
In vivo mice experiment
Five-week-old male immune deficient nude mice (BALB/c nu) were purchased from the animal center of the Chinese Academy of Medical Sciences, fed at the laboratory animal center of Soowhow University. U266 cells (4 × 106/mouse) were subcutaneously injected in the flanks of 5-week-old nude female mice. Mice were then randomly distributed into four groups (3/group). After the formation of palpable tumors, treatment was commenced. Tumor volume was calculated using a caliper. Mice were euthanized 28 days after treatment in four groups. Tumor volume was evaluated from the first day of treatment. Mice were treated in a humane manner in compliance with ARRIVE guidelines, animal experiment was approved by the Ethical Committee of Animal Study.
Biostatistics
Unless otherwise specified, the data presented are the mean ± SD. GraphPad Prism 9 (GraphPad) software was utilized for statistical analysis. For group comparisons, the Student’s t-test (unpaired and two-tailed) was employed (*P < 0.05, **P < 0.01, ***P < 0.005), and exact P values are stated in the source data for each figure panel.
| Results | ▴Top |
Bortezomib decreased MM cell viability, proliferation rate and induced apoptosis
MM cell lines U266 and RPMI8226 were incubated with bortezomib at indicated concentrations for 24 h. CCK8 was used to calculate the IC50 (half maximal inhibitory concentration) of bortezomib for U266 and RPIM8226 at 24 h. The IC50 of bortezomib for U266 and RPMI8226 were 12.05 nM and 16.30 nM. Bortezomib decreased cell viability of both cell lines in a dose-dependent manner (Fig. 1a). We next determined the effect of bortezomib on cell proliferation by measuring BrdU incorporation. The cell proliferation rate of U266 and RPIM8226 was tested with BrdU incubation for 2 h after bortezomib treatment for 24 h,bortezomib generated a dose-dependent increase in absorbance (Fig. 1b). The results indicated that bortezomib alone increased BrdU incorporation. To determine the cell death manner, MM cells were incubated with bortezomib for 24 h and were added with caspase 3 or TUNEL 3 h before detection. The relative percentage of cells undergoing apoptosis was determined by caspase 3 and TUNEL staining (Fig. 1c). Bortezomib induced a dose-dependent increase of cleaved caspase 3-positive (Fig. 1d), and TUNEL-positive (Fig. 1e) cells in U266. These results indicated that bortezomib decreased the cell viability of MM cell lines through caspase 3 mediated apoptosis.
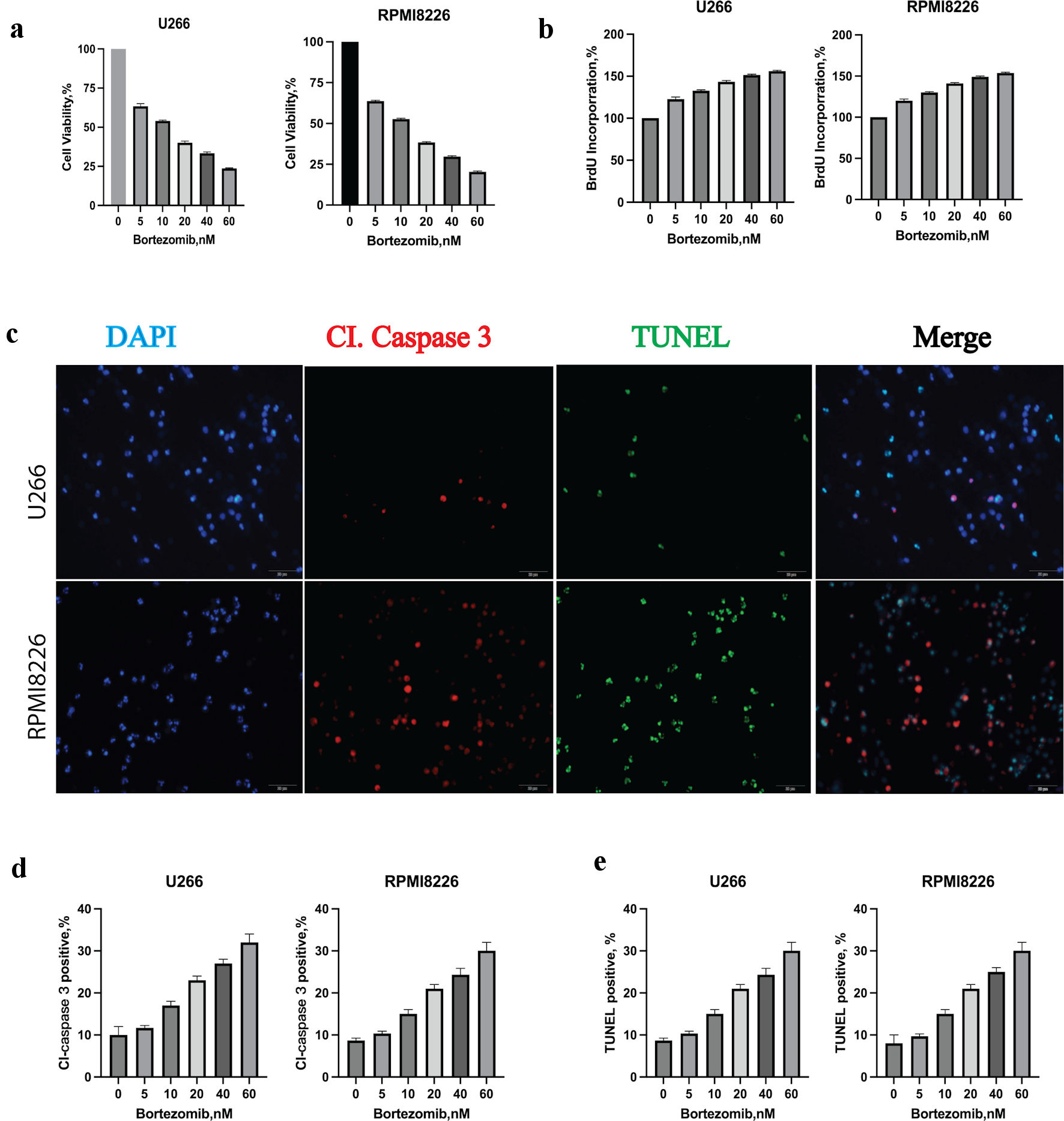 Click for large image | Figure 1. Bortezomib decreased multiple myeloma (MM) cell viability, proliferation rate and induced apoptosis. (a) Bortezomib decreased the viability of U266 and RPMI8226 at indicated concentrations with the CCK8 assay. Error bars represent SD values determined from triplicate measurements. (b) Bortezomib decreased cell proliferation of U266 and RPMI8226. Cells were incubated with bortezomib at indicated concentrations for 24 h and BrdU for 2 h. BrdU incorporation was determined relative to untreated cells. Error bars represent SD values determined from triplicate measurements. (c) Bortezomib induced apoptosis in U266 cell line. U266 cells were incubated with bortezomib, caspase 3 and TUNEL for 24 h. MM cells were then visualized by staining and confocal microscopy for caspase 3 (red) and TUNEL (green) to label apoptosis. (d) The relative amount of caspase 3 (red) fluorescence was determined using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Error bars represent SD values from triplicate measurements. (e) The relative amount of TUNEL (green) fluorescence was determined using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Error bars represent SD values from triplicate measurements. CCK8: cell counting kit-8; SD: standard deviation; DAPI: 4’,6-diamidino-2-phenylindole; TUNEL: terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling. |
SR9009 decreased MM cell viability, proliferation rate and induced apoptosis
CCK8 was used to calculate the IC50 of SR9009 at 48 h for U266 and RPIM8226. The IC50 of SR9009 for U266 and RPMI8226 were 22.10 µM and 29.35 µM. Similarly, SR9009 decreased cell viability of both cell lines in a dose-dependent manner (Fig. 2a). The cell proliferation rate of U266 and RPIM8226 was tested with BrdU labeling after incubation with SR9009 for 48 h (Fig. 2b). These results showed that SR9009 could slow down MM cell proliferation. To determine the cell death manner, MM cell lines were incubated with SR9009 for 48 h and were added with caspase 3 and TUNEL 3 h before test. The relative percent of cells undergoing apoptosis was determined by caspase 3 and TUNEL staining (Fig. 2c). SR9009 induced a dose-dependent increase of cleaved caspase 3-positive (Fig. 2d) and TUNEL-positive (Fig. 2e) cells in U266 and RPIM8226. Together, these results indicated that SR9009 decreased MM cell viability through caspase 3-mediated apoptosis. These results showed that both bortezomib and SR9009 could induce apoptosis in MM. But we wanted to know whether there were any differences.
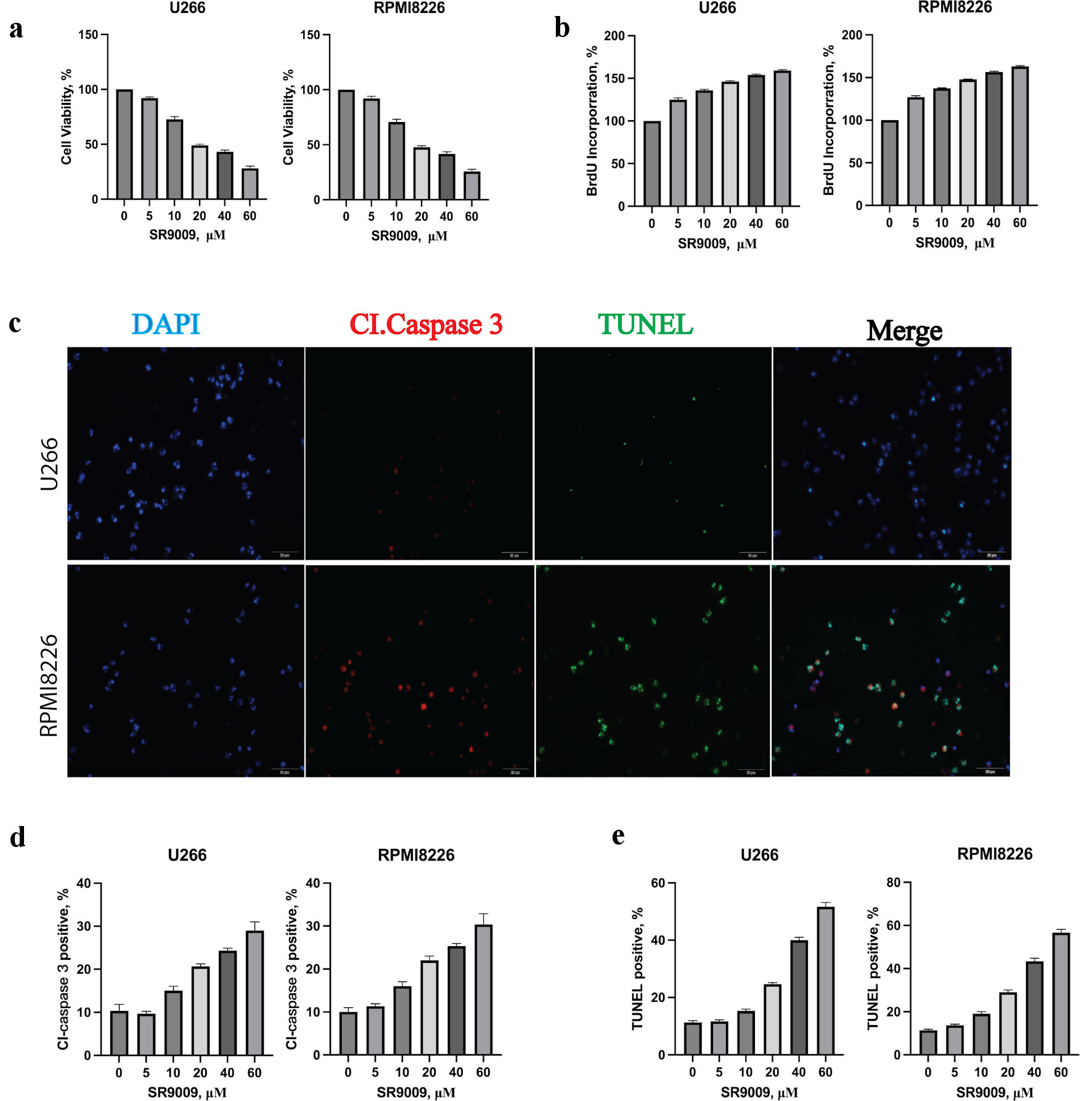 Click for large image | Figure 2. Circadian clock REV-ERB agonist SR9009 decreased multiple myeloma (MM) viability, proliferation rate and induced an apoptotic response in MM cells. (a) SR9009 decreased the viability of U266 and RPMI8226 at indicated concentrations with the CCK8 assay. Error bars represent SD values determined from triplicate measurements. (b) SR9009 decreased the proliferation rate of U266 and RPMI8226. Cells were incubated with SR9009 at indicated concentrations for 48 h and BrdU for 2 h. BrdU incorporation was determined relative to untreated cells. Error bars represent SD values determined from triplicate measurements. (c) SR9009 induced apoptosis in U266 cell line. U266 cells were incubated with SR9009, caspase 3 and TUNEL for 24 h. Cells were then visualized by confocal microscopy for caspase 3 (red) and TUNEL (green) to label apoptosis. (d) The relative amount of caspase 3 (red) fluorescence was determined using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Error bars represent SD values from triplicate measurements. (e) The relative amount of TUNEL (green) fluorescence was determined using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Error bars represent SD values from triplicate measurements. CCK8: cell counting kit-8; SD: standard deviation; DAPI: 4’,6-diamidino-2-phenylindole; TUNEL: terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling. |
Bortezomib increased GRP78 and autophagy, SR9009 decreased GRP78 and blocked autophagy
To determine whether bortezomib would induce a cell-protective increase in GRP78 and autophagy, we analyzed GRP78 and LC3 protein modifications using immunoblot. As anticipated, bortezomib increased GRP78 and caused a dose-dependent conversion of LC3I to LC3II (Fig. 3a). To determine the effect of bortezomib on GRP78, LC3 levels in U226 cell lysates, quantitative analysis of GRP78, LC3, and GAPDH was performed using ImageJ (Fig. 3b). As anticipated, SR9009 upregulated the protein levels of REV-ERBα and REV-ERBβ since it was an agonist for both REV-ERBα and REV-ERBβ. In contrast to bortezomib, we observed that SR9009 inhibited autophagy induction and decreased GRP78 protein levels. Since GRP78 was necessary for ER integrity and function as well as autophagy, we hypothesized that the GRP78-dependent autophagy process could also be inhibited. As anticipated, instead of observing a greater conversion of LC3A to LC3B, we observed that both LC3A and LC3B decreased proportionally; this result suggested that SR9009 was distinct from conventional autophagy inhibitors such as 3-MA or chloroquine (Fig. 3c). Microtubule-associated protein light chain I (LC3A) is covalently linked to phosphatidylethanolamine and bound to the autophagosome membrane, where it recruits cargo proteins for degradation, through autophagosome formation. The process of lipidation transforms cytoplasmic LC3A into the membrane-bound active form LC3B. Incubation with bortezomib significantly increased the conversion of LC3A to LC3B, as determined by immunoblot. By reducing the levels of LC3A and LC3B proportionally, SR9009 blocked autophagy induction from the initiation stage. ImageJ was used for quantitative analysis of GRP78, LC3 and GAPDH (Fig. 3d). As both NR1D1 and NR1D2 agonist, SR9009 upregulated NR1D1 and NR1D2 at the mRNA and protein levels (Fig. 3e, f). These results indicated that SR9009 acted as an agonist for REV-ERBs and induced MM to undergo apoptosis by inhibiting autophagy.
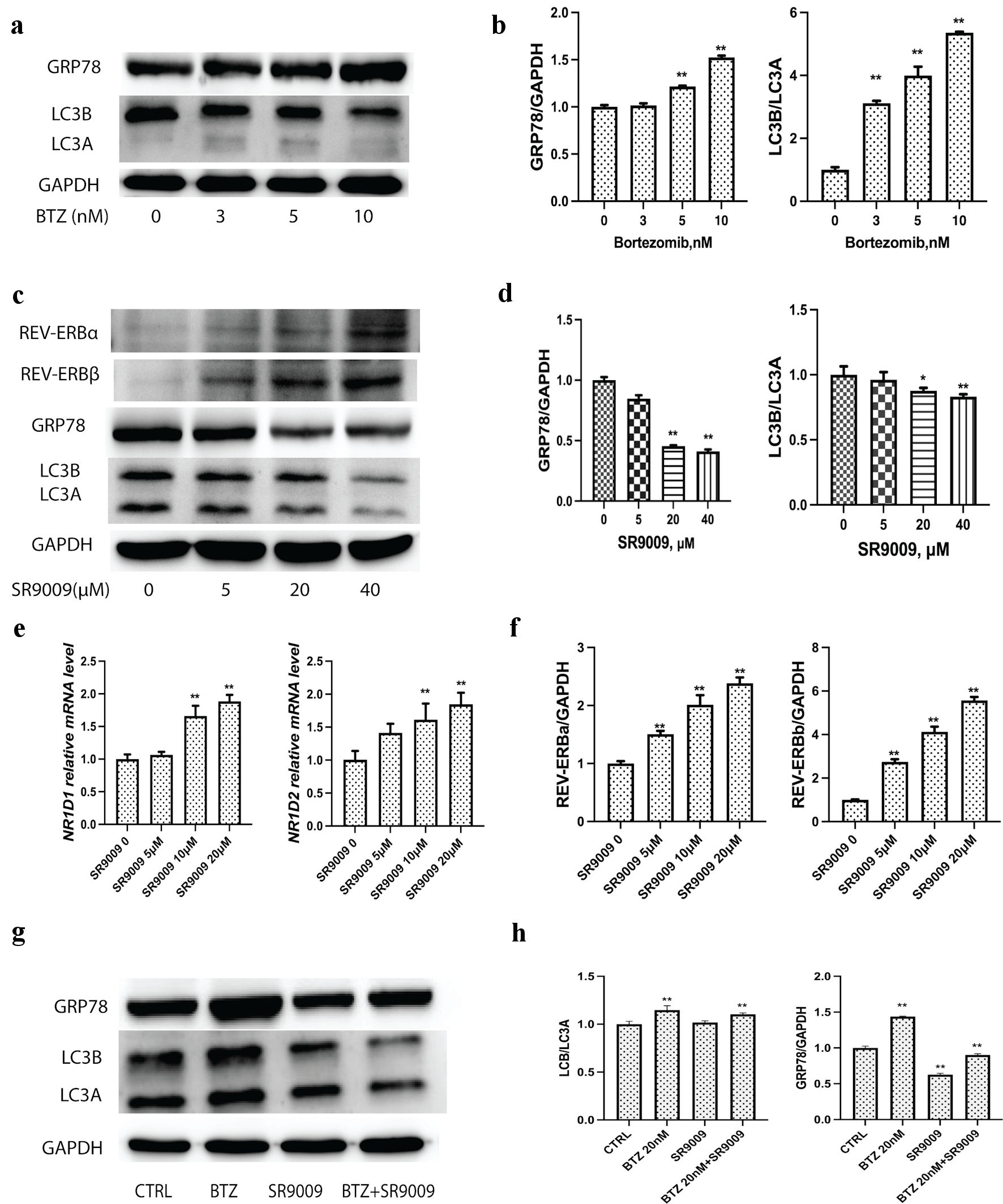 Click for large image | Figure 3. Bortezomib upregulated GRP78 and autophagy, while SRR9009 downregulated GRP78 and blocked autophagy induction. (a) Representative immunoblots of GRP78, LC3 and GAPDH to determine the effect of bortezomib treatment on GRP78, LC3 levels in U226 cell lysates. Cells were treated with bortezomib at indicated concentrations for 24 h; lysates were prepared and probed using a GRP78-specific and LC3-specific antibody. (b) Quantitative analysis of GRP78, LC3 and GAPDH were measured. Gray intensity of GRP78, LC3II/ LC3I from the immunoblot results were measured with ImageJ to determine the effect of bortezomib treatment on GRP78, LC3 levels in U226 cell lysates. (c) SR9009 upregulated REV-ERBα and REV-ERBβ at the protein level. SR9009 downregulated the GRP78 protein level and blocked autophagy in U226 cells by immunoblot. Cells were incubated with bortezomib with indicated concentrations. (d) Quantitative analysis of GRP78, LC3 and GAPDH to determine the effect of SR9009 treatment on GRP78, LC3 levels in U226 cell lysates. Gray intensity of GRP78, LC3II/LC3I from immunoblot results were measured with ImageJ. (e) As NR1D1 and NR1D2 agonist, SR9009 upregulated NR1D1 and NR1D2 at the mRNA level. (f) Quantitative analysis of REV-ERBα, REV-ERBβ. Gray intensity of the immunoblot results were measured with ImageJ. (g) Bortezomib upregulated GRP78, LC3I and LC3II, SR9009 downregulated GRP78, LC3I and LC3II. Combination of SR9009 could prevent bortezomib-induced GRP78 and LC3 upregulation in MM. (h) Quantitative analysis of GRP78 and LC3 were measured with ImageJ for immunoblot results. GRP78: glucose-regulated protein 78; GAPDH: glyceraldehyde-3-phosphate dehydrogenase (GAPDH); MM: multiple myeloma; BTZ: bortezomib. |
The accumulation of misfolded proteins in the ER lumen initiates the downstream autophagy process through GRP78. Since bortezomib treatment increased the GRP78 level in U266 lysate, while SR9009 significantly reduced the GRP78 level in U226 lysate. We hypothesized that co-incubation of SR9009 with bortezomib could prevent the elevation of GRP78 and the process of autophagy. Immunoblot results indicated that adding SR9009 can partially prevent the increase of GRP78 and autophagy induction induced by bortezomib (Fig. 3g). Quantitative analysis of GRP78 and LC3 were measured with ImageJ (Fig. 3h).
Bortezomib increased autophagy LC3, while SR9009 decreased autophagy LC3
MM cell lines U266 and RPMI8226 were transfected with adenovirus with double-labeled autophagy effecter LC3. Mechanically, when the autophagosome fused with the lysosome, the green GFP fluorescence is quenched, and only red fluorescence can be detected.
By quantifying green fluorescently labeled autophagosomes, bortezomib alone upregulates autophagosomes and SR9009 downregulated LC3 fluorescence and autophagosomes (Fig. 4a). SR9009 partially prevented the formation of autophagosomes induced by bortezomib (Fig. 4a). Quantitative analysis for the confocal autophagy flux results was presented (Fig. 4c). The confocal results were in accordance with the immunoblot results, SR9009 downregulated autophagosomes and autolysosomes, while bortezomib unregulated autophagosomes and autolysosomes. Chloroquine inhibits the late stage of autophagy by blocking the fusion of autophagosomes to lysosomes, leading to the accumulation of autophagosomes. Different from chloroquine, the agonist of REV-ERBs SR9009 reduced the number of autophagosomes. Both bortezomib and SR9009 induced apoptosis in MM. MM cell lines were treated with bortezomib at the indicated concentrations for 24 h, and the relative percent of cells undergoing apoptosis was determined by annexin-V-FITC staining and flow cytometry (Fig. 4b). Both bortezomib and SR9009 generated an increase in annexin-positive cells to indicate that both bortezomib and SR9009 could mediate MM apoptosis. Quantitative analysis of flowcytometry apoptosis assay was shown in Figure 4d. These results indicated that bortezomib induced enhanced autophagy flux, while SR9009 blocked the autophagy flux. SR9009 had synergistic effect with bortezomib by increasing the apoptosis.
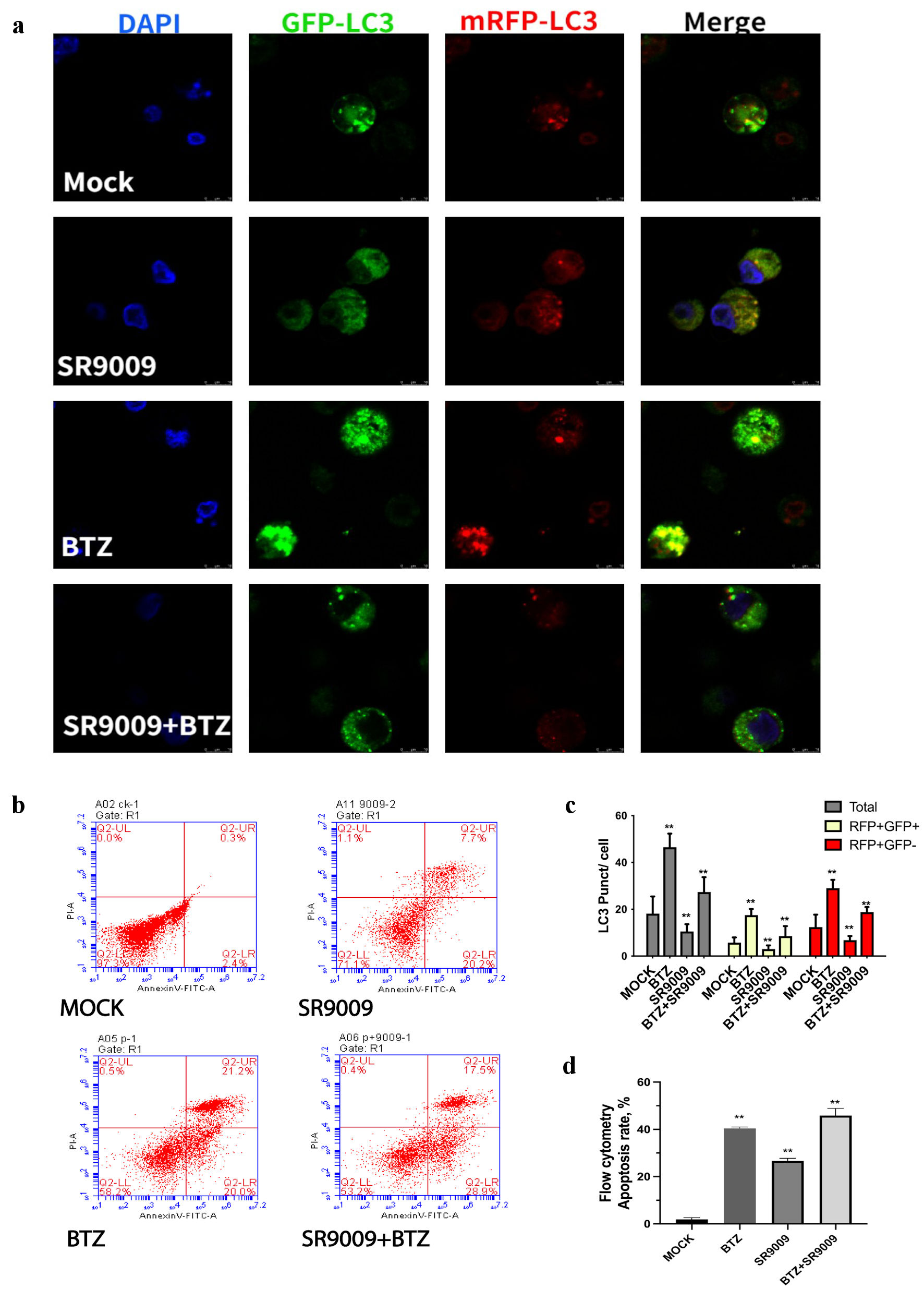 Click for large image | Figure 4. SR9009 decreased LC3, bortezomib increased LC3. (a) Representative images of LC3 puncta, autophagosomes were detected with confocal microscopy. Cultured U266 were transduced with dual-fluorescence labeled adenovirus-tf-LC3 and then incubated with mock, SR9009, bortezomib, and both. Representative images of green GFP-LC3 puncta indicated auto-phagosomes, whereas red mRFP-LC3 puncta indicated auto-lysosomes. SR9009 downregulated auto-phagosomes and auto-lysosomes, bortezomib upregulated auto-phagosomes and auto-lysosomes. (b) Multiple myeloma (MM) cell lines were treated with mock, bortezomib, SR9009, bortezomib + SR9009 at the indicated concentrations for 24 h and the relative percent of cells undergoing apoptosis was determined by annexin V-fluorescein isothiocyanate (V-FITC) staining and flow cytometry. (c) The relative amount of green and red fluorescence was determined using ImageJ software (National Institutes of Health, Bethesda, MD, USA). The bar graph indicated mean numbers of GFP-LC3 and mRFP-LC3 per cell obtained from three experiments. (d) Quantitative analysis of flowcytometry apoptosis assay. Both bortezomib and SR9009 generated an increase in annexin-positive cells. DAPI: 4’,6-diamidino-2-phenylindole; BTZ: bortezomib. |
SR9009 downregulated core autophagy genes, blocked de novo lipogenesis and enhanced the anti-myeloma effect of bortezomib in vivo
In untreated U266 cells, GRP78 levels were not high, but the fluorescence level of GRP78 increased significantly after bortezomib incubation (Fig. 5a). SR9009 decreased the fluorescence intensity of GRP78 (Fig. 5a). Adding SR9009 could partially inhibit the GRP78 elevation induced by bortezomib (Fig. 5a). The GRP78 immunofluorescence intensity was measured with quantitative analysis (Fig. 5b). The confocal GRP78 protein results were consistent with the immunoblot results in Figure 3c, supporting the results that SR9009 could inhibit the ER stress sensor GRP78 and the autophagy process.
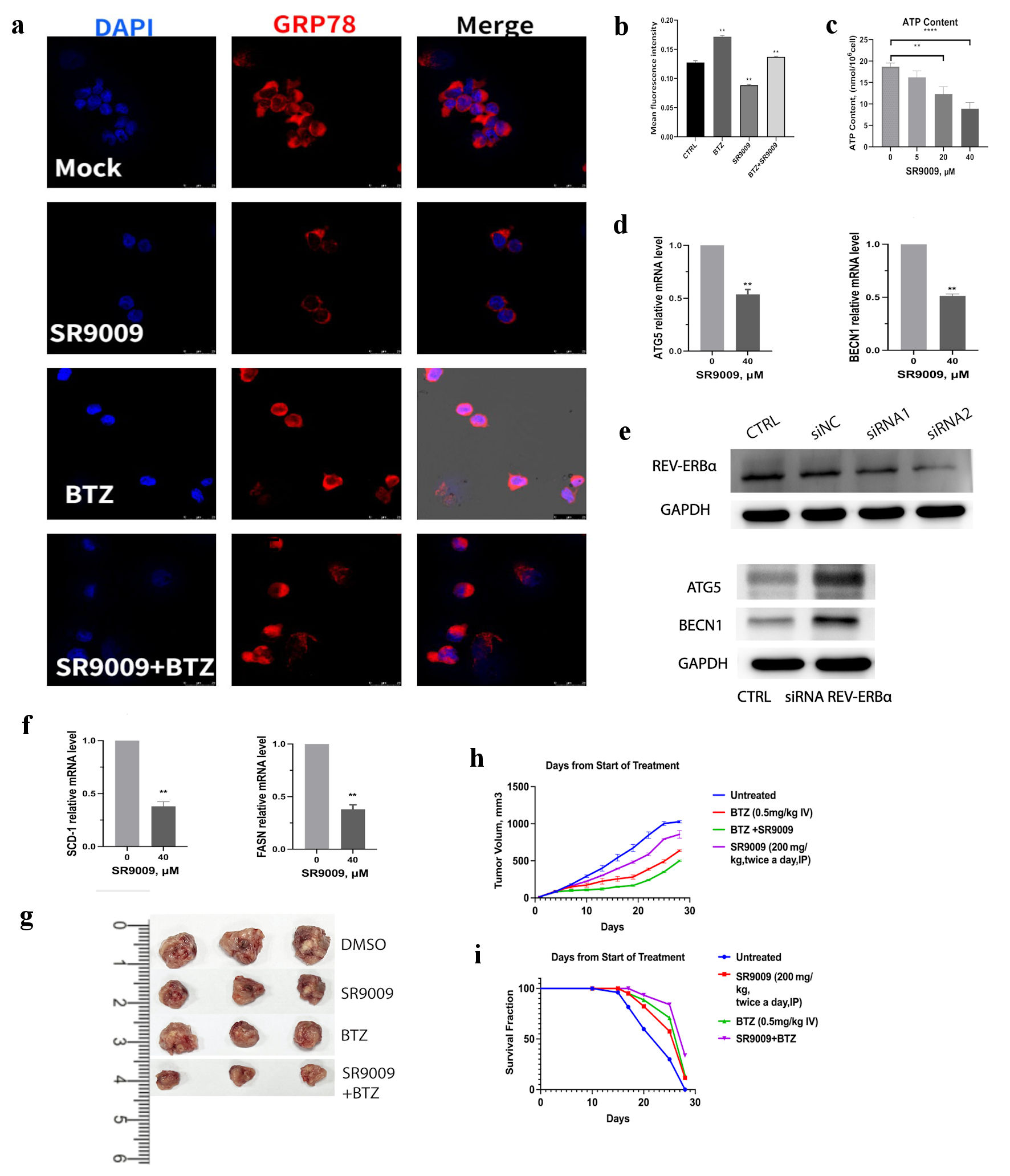 Click for large image | Figure 5. SR9009 decreased core autophagy genes, blocked de novo lipogenesis and showed synergistic effect with bortezomib in vivo. (a) Multiple myeloma (MM) was incubated with human GRP78-specific monoclonal antibody and observed with confocal assay for GRP78 protein in U266 cell lines. U266 cells were treated with mock, SR9009 (40 µM), bortezomib (5 nM), bortezomib (5 nM) with SR9009. (b) Quantitative analysis of GRP78 immunofluorescence intensity were measured with ImageJ. (c) REV-ERBs agonists SR9009 decreased mitochondria ATP production in a dose-dependent manner. Statistically significant differences were indicated by (**P < 0.01, ***P < 0.001). (d) SR9009 downregulated core autophagy genes ATG5 and BENC1 at the mRNA level with qRT-PCR. (e) Knockdown of REV-ERBα by siRNA, siRNA2 was used for the next experiments. REV-ERBα knockdown leads to upregulation of ATG5 and BENC1 at the protein level with immunoblot results. (f) SR9009 downregulated two essential enzymes for de novo lipogenesis SCD1 and FASN at the mRNA level with qRT-PCR. (g) Representative tumor images in four groups. U266 cells (4 × 106/mouse) were subcutaneously injected in the flanks of 5-week-old nonobese diabetic/severe combined immunodeficient (NOD/SCID) female mice. Mice were then randomly distributed into four groups (3/group). After the formation of palpable tumors, treatment was commenced. Mice received intravenous injection of either vehicle (phosphate-buffered saline containing 10% dimethyl sulfoxide (DMSO)) or bortezomib (0.5 mg/kg in phosphate-buffered saline, 10% DMSO) administered with or without SR9009 (600 µg/mL). Bortezomib was administered intravenously with or without SR9009 for 5 days. (h) Tumor volume was calculated using a caliper. Mice were euthanized 28 days after treatment. Tumor volume was evaluated from the first day of treatment. Shown is the average of replicate measurements. (i) Kaplan-Meier survival curves after treated with either vehicle, SR9009, bortezomib or both. SCD1: stearoyl-CoA desaturase 1; FASN: fatty acid synthase; qRT-PCR: quantitative real-time polymerase chain reaction; ATP: adenosine triphosphate; DAPI: 4’,6-diamidino-2-phenylindole; BTZ: bortezomib. |
Next, we sought to determine whether REV-ERBs agonist SR9009 affect mitochondria functions. Normally, mitochondria are regulated by quality control mechanisms, such as fission and fusion, biogenesis, and autophagy [23, 24]. Mitochondria are the major organelle of cellular ATP production, but when they malfunction, they are also the major source of oxidative stress which finally triggers apoptosis or necrosis [25]. Mitophagy delivers misfolded and malfunctioning mitochondria for recycling [26, 27]. We proposed that mitochondria functions might be affected by SR9009’s blockade of autophagy. Results showed that ATP production was decreased with SR9009 incubation (Fig. 5c).
We wanted to gain further mechanistic insights whether the core autophagy genes were also affected by SR9009, we noticed a decrease in the expression of ATG5, and BECN1 mRNA level (Fig. 5d). Since SR9009 could inhibit ATG5 and BECN1, we hypothesized that ATG5 and BECN1 expression might be controlled by circadian clock. So, we knockdown REV-ERB by siRNA, siRNA2 was used for the next experiments (Fig. 5e). Immunoblot results showed that REV-ERBα knockdown lead to upregulation of ATG5 and BECN1 (Fig. 5e). These results suggested that the core autophagy genes ATG5 and BECN1 were circadian clock controlled.
Autophagy blockade impaired the cellular fitness by cutting off nutrient supply, especially for cancer cells which have high metabolic stress and high demand for nutrients. Lipid is necessary for autophagy functions, we hypothesized that SR9009 might play a role in the fatty acid synthesis process. Cancer cells are highly dependent on de novo lipogenesis [28, 29]; treatments for specific lipogenic enzyme inhibitors are currently underway, including fatty acid synthase (FASN) and stearoyl-CoA desaturase 1 (SCD1) as targets. SR9009 reduced the mRNA level of two key rate-limiting fatty acid synthesis enzymes, which function in de novo lipogenesis (Fig. 5f). Taken together, these results indicated that circadian clock REV-ERBs agonist SR9009 reduced the ER stress sensor GRP78, inhibited the core autophagy genes ATG5, BECN1 and blocked autophagy at the initiation stage. SR9009 also inhibited the genes FASN and SCD1 (two essential enzymes for de novo lipogenesis at the mRNA level). All the above discussed mechanisms contribute to MM cell apoptosis.
Mice xenograft models were used to evaluate the in vivo effect of SR9009 and bortezomib. SR9009 was administered twice per day by intraperitoneal (ip) injection at the indicated concentrations for 1 week. Bortezomib was administered intravenously (0.5 mg/kg in phosphate-buffered saline, 10% dimethyl sulfoxide (DMSO)) with or without SR9009 for 5 days. All mice in this study were kept according to guidelines approved by the Animal Care and Use Committee of Soochow University. Mice were killed 1 h after the final treatment. We monitored the tumor burden and volume (Fig. 5g, h) and mice’s overall survival time (Fig. 5i). Bortezomib alone reduced tumor volume relative to the untreated group or SR9009 group. The effect of bortezomib was enhanced with SR9009. Median survival was 18 days in the control group, 20 days in the SR9009-treated group, 23 days in the bortezomib-treated group and 27 days in the bortezomib + SR9009 group (Fig. 5i). The results suggested that SR9009 augmented the antitumor effect of bortezomib, slowed down tumor growth and had survival benefit.
| Discussion | ▴Top |
In this study, we investigated how disrupted circadian clock affects the drug resistance of proteasome inhibitors and provide a rational to take advantage of this vulnerability. Although both bortezomib and SR9009 decreased cell viability and induced apoptosis in MM cell lines. We observed many great differences in their actions: bortezomib elicited an ER stress by increasing GRP78 level by western blotting, and confocal results. And GRP78 functioned as a protective mechanism for MM survival [30, 31], during which the UPR regulator GRP78 facilitated autophagy and lipogenesis [12]. On the contrary, SR9009 decreased GRP78 and LC3. SR9009 had synergistic role with bortezomib by preventing the upregulation of GRP78 and autophagy induction.
Since lipidation and lipogenesis were coupled with autophagy process [32], we hypothesized that the lipogenesis process might be influenced by SR9009. By comparing the changes of two essential enzymes for de novo lipogenesis: FASN and SCD1, we found that SR9009 blocked not only the autophagy initiation process, but also inhibited the two essential enzymes FASN and SCD1 at the mRNA level. Taken together, these results indicated that SR9009 could overcome the side effect of proteasome inhibitors, and combining SR9009 would be a very good and rational strategy for MM treatment. Mechanically, circadian clock agonist SR9009 inhibited GRP78 and autophagy, and induced apoptosis in MM. Moreover, SR9009 inhibited the core autophagy gene ATG5 and BECN1, and two essential enzymes for de novo lipogenesis FASN and SCD1.
By disrupting the mitochondria homeostasis and decreased ATP production, SR9009 disrupted MM energy homeostasis under stress conditions. Mice xenograft models were used to evaluate the safety and efficacy of SR9009 alone and in combination with bortezomib. SR9009 alone and bortezomib alone could decrease MM tumor growth; the combination group had the slowest tumor growth and the maximum mice survival time. Our results solved the toughest issue with proteasome inhibitors; by preventing ER stress and protective autophagy in MM cell lines, SR9009 had synergistic antitumor effect with proteasome inhibitors.
Proteasome inhibitors inevitably induce a terminal UPR and tumor protective autophagy for survival. However, the metabolic mechanisms for drug resistance are not well illustrated. Here, we found that circadian clock component REV-ERBs agonist SR9009 could block autophagy and lipogenesis process and showed synergistic effect with proteasome inhibitors. Mice xenograft models showed safety and efficacy. Mechanically, SR9009 blocked autophagy at the initiation stage and downregulated the lipogenesis coupled with autophagy process. SR9009 downregulated the core autophagy genes ATG5 and BECN1, and two essential enzymes for de novo lipogenesis: FASN and SCD1. Our results provide a deep insight into the metabolic mechanisms for proteasome inhibitor-induced drug resistance, and illustrated well how disrupted tumor circadian clock could provide a targeting vulnerability in cancer therapy.
Acknowledgments
We are grateful to the members of our laboratory for discussions and technical help.
Financial Disclosure
This research was supported by fundings from Natural Science Foundation of Suqian Science and Technology Bureau (K201903, Z2018076, Z2018213 and Z2022065), Jiangsu Science and Technology Association (JSTJ-2022-004).
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
Not applicable.
Author Contributions
Rui Wang conceived and designed the study, conducted experiments, analyzed the data, and wrote the manuscript. Mei Mei Ma and Shu Ling Liu helped with conducting experiments and revised the manuscript. Quan Quan Guo and Xiao Hong Shi contributed to discussion and manuscript editing and revising. All authors discussed the results and commented on the manuscript.
Data Availability
Any inquiries regarding supporting data availability of this study should be directed to the corresponding author.
| References | ▴Top |
- Chauhan D, Singh A, Brahmandam M, Podar K, Hideshima T, Richardson P, Munshi N, et al. Combination of proteasome inhibitors bortezomib and NPI-0052 trigger in vivo synergistic cytotoxicity in multiple myeloma. Blood. 2008;111(3):1654-1664.
doi pubmed pmc - Laubach JP, Mahindra A, Mitsiades CS, Schlossman RL, Munshi NC, Ghobrial IM, Carreau N, et al. The use of novel agents in the treatment of relapsed and refractory multiple myeloma. Leukemia. 2009;23(12):2222-2232.
doi pubmed pmc - Obeng EA, Carlson LM, Gutman DM, Harrington WJ, Jr., Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107(12):4907-4916.
doi pubmed pmc - Rasche L, Menoret E, Dubljevic V, Menu E, Vanderkerken K, Lapa C, Steinbrunn T, et al. A GRP78-directed monoclonal antibody recaptures response in refractory multiple myeloma with extramedullary involvement. Clin Cancer Res. 2016;22(17):4341-4349.
doi pubmed - Abdel Malek MA, Jagannathan S, Malek E, Sayed DM, Elgammal SA, Abd El-Azeem HG, Thabet NM, et al. Molecular chaperone GRP78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget. 2015;6(5):3098-3110.
doi pubmed pmc - Jagannathan S, Abdel-Malek MA, Malek E, Vad N, Latif T, Anderson KC, Driscoll JJ. Pharmacologic screens reveal metformin that suppresses GRP78-dependent autophagy to enhance the anti-myeloma effect of bortezomib. Leukemia. 2015;29(11):2184-2191.
doi pubmed pmc - Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, Quarato G, et al. LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell. 2018;175(2):429-441.e416.
doi pubmed pmc - Wan J, Weiss E, Ben Mkaddem S, Mabire M, Choinier PM, Thibault-Sogorb T, Hegde P, et al. LC3-associated phagocytosis in myeloid cells, a fireman that restrains inflammation and liver fibrosis, via immunoreceptor inhibitory signaling. Autophagy. 2020;16(8):1526-1528.
doi pubmed pmc - Lee HC, Shah JJ, Orlowski RZ. Novel approaches to treatment of double-refractory multiple myeloma. Am Soc Clin Oncol Educ Book. 2013;2013:302-306.
doi pubmed pmc - Dong D, Stapleton C, Luo B, Xiong S, Ye W, Zhang Y, Jhaveri N, et al. A critical role for GRP78/BiP in the tumor microenvironment for neovascularization during tumor growth and metastasis. Cancer Res. 2011;71(8):2848-2857.
doi pubmed pmc - Fu Y, Lee AS. Glucose regulated proteins in cancer progression, drug resistance and immunotherapy. Cancer Biol Ther. 2006;5(7):741-744.
doi pubmed - Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008;15(9):1460-1471.
doi pubmed pmc - Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 2007;67(8):3496-3499.
doi pubmed - Wang Q, He Z, Zhang J, Wang Y, Wang T, Tong S, Wang L, et al. Overexpression of endoplasmic reticulum molecular chaperone GRP94 and GRP78 in human lung cancer tissues and its significance. Cancer Detect Prev. 2005;29(6):544-551.
doi pubmed - Lee HK, Xiang C, Cazacu S, Finniss S, Kazimirsky G, Lemke N, Lehman NL, et al. GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro Oncol. 2008;10(3):236-243.
doi pubmed pmc - Zhang J, Jiang Y, Jia Z, Li Q, Gong W, Wang L, Wei D, et al. Association of elevated GRP78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin Exp Metastasis. 2006;23(7-8):401-410.
doi pubmed - Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8(8):627-644.
doi pubmed pmc - Fu L, Lee CC. The circadian clock: pacemaker and tumour suppressor. Nat Rev Cancer. 2003;3(5):350-361.
doi pubmed - Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.
doi pubmed - Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, Chong LW, et al. Regulation of circadian behaviour and metabolism by REV-ERB-alpha and REV-ERB-beta. Nature. 2012;485(7396):123-127.
doi pubmed pmc - Bugge A, Feng D, Everett LJ, Briggs ER, Mullican SE, Wang F, Jager J, et al. Rev-erbalpha and Rev-erbbeta coordinately protect the circadian clock and normal metabolic function. Genes Dev. 2012;26(7):657-667.
doi pubmed pmc - Hastings MH, Smyllie NJ, Patton AP. Molecular-genetic manipulation of the suprachiasmatic nucleus circadian clock. J Mol Biol. 2020;432(12):3639-3660.
doi pubmed - Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell. 2016;166(3):555-566.
doi pubmed pmc - Abate M, Festa A, Falco M, Lombardi A, Luce A, Grimaldi A, Zappavigna S, et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin Cell Dev Biol. 2020;98:139-153.
doi pubmed - Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. 2020;21(4):204-224.
doi pubmed - Yoo SM, Jung YK. A Molecular Approach to Mitophagy and Mitochondrial Dynamics. Mol Cells. 2018;41(1):18-26.
doi pubmed pmc - Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021;40(3):e104705.
doi pubmed pmc - Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond). 2018;38(1):27.
doi pubmed pmc - Bartolacci C, Andreani C, Vale G, Berto S, Melegari M, Crouch AC, Baluya DL, et al. Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer. Nat Commun. 2022;13(1):4327.
doi pubmed pmc - Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26(24):9220-9231.
doi pubmed pmc - Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, et al. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem. 2007;282(7):4702-4710.
doi pubmed - Sekar M, Thirumurugan K. Autophagy: a molecular switch to regulate adipogenesis and lipolysis. Mol Cell Biochem. 2022;477(3):727-742.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.