

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website http://www.wjon.org |
Case Report
Volume 2, Number 1, February 2011, pages 41-44
Increased Incidence of Tumors With the IKBKAP Gene Mutation?A Case Report and Review of the Literature
Marianna Shvartsbeyna, d, Amy Rapkiewicza, Felicia Axelrodb, c, Horacio Kaufmannb
aDepartment of Pathology, New York University, New York, USA
bDepartment of Pediatrics, New York University, New York, USA
cDepartment of Neurology, New York University, New York, USA
dCorresponding author: Marianna Shvartsbeyn, Department of Pathology, New York University, 522 First Avenue, Smilow, 301C, New York, NY 10016, USA
Manuscript accepted for publication January 27, 2011
Short title: IKBKAP Gene Mutation in Tumors
doi: https://doi.org/10.4021/wjon278w
| Abstract | ▴Top |
An increased incidence of neoplasia was recently reported in patients with familial dysautonomia. This suggests that, in addition to its role in neuronal development, the IKBKAP gene may also influence DNA repair. Here we report the case of a 28-year-old male with familial dysautonomia who was found to have neoplastic lesions detected post mortem as incidental findings. This case indicates that the prevalence of tumorgenesis within this population may be underestimated.
Keywords: Familial dysautonomia; IKBKAP; Tumorgenesis
| Introduction | ▴Top |
Familial dysautonomia, also known as Riley-Day syndrome and hereditary sensory and autonomic neuropathy type III, is a rare autosomal recessive disorder. Over 99.5% of cases are homozygous for a point mutation in the 5’ splice site of the IKBKAP gene located on the long arm of chromosome 9 (9q31), which encodes IkB kinase complex associated protein (IKAP) [1, 2]. The carrier rate among Askenazi Jews is approximately 1 in 27. The disorder is characterized by sensory and autonomic dysfunction as a result of an abnormality in the development in the sensory and autonomic neuronal pathways [3].
The protein IKAP is predominantly present in the cytoplasm where it interacts with other proteins involved in cellular migration, adhesion and actin cytoskeleton organization [4]. The IKBKAP gene sequence bears a striking similarity to the ELP1 gene, which encodes a subunit of a complex that binds to RNA polymerase II and is required for the activation and transcriptional elongation of a large number of genes [5]. Similar to ELP1, IKAP may be a part of the RNA polymerase II elongation complex and might play a role in expression of the genes involved in neural migration, differentiation and vascular development in utero [4, 6-9]. Interestingly, in vitro experiments showed that fibroblasts from familial dysautonomia patients had increased susceptibility to DNA damage after exposure to N-methyl-N’-nitro-N-nitrosoguanidine [10]. This finding suggests an inability to effectively repair damaged DNA and may explain the increased incidence of tumorgenesis in these patients.
| Case Report | ▴Top |
The decedent was a 28-year-old Ashkenazi Jewish man diagnosed with familial dysautonomia by genetic testing. At 6 weeks of age he presented with hypothermia and severe feeding problems. He had alacrima and other characteristic features of familial dysautonomia including unstable blood pressure and gastrointestinal dysfunction with oral incoordination and gastroesophageal reflux. He underwent a Nissen fundoplication and gastrostomy at age 2, suffered three episodes of intestinal obstruction, requiring bowel resection, and multiple bouts of pancreatitis and cholecystitis, persistent esophageal dysmotility leading to multiple episodes of aspiration pneumonia. Cinesophagram demonstrated marked esophageal dilation with poor peristaltic wave and narrowed spastic esophagogastric junction. He had kyphoscoliosis and underwent spine fusion at the age of ten; he also had equinovarus deformity and osteoporosis with multiple bone fractures. He was classified as learning disabled. He suffered several seizures associated with hyponatremia and hypoxia. Because of obstructive sleep apnea and nocturnal oxygen desaturations and hypercapnia that persisted after tonsillectomy and adenoidectomy, non-invasive positive pressure respiration was recommended but refused. In the months preceding his death, his condition was stable and there was no obvious change in his status on the evening that preceded his unexpected death during sleep.
The salient findings at autopsy were the esophagus distended with partially undigested food and evidence of aspiration pneumonia with associated foreign body giant cell reaction on the lung sections. Taken together in a context of swallowing difficulty and esophageal dysmotility, these findings were consistent with aspiration as a cause of death.
The unexpected findings were multiple well-circumscribed unencapsulated, pale yellow tumors in the subcapsular portions of both kidneys ranging from 0.2 to 0.4 cm in greatest dimension (Fig. 1). H&E sections revealed densely packed papillae or tubules lined by small, regular, cuboidal cells with rounded, uniform nuclei that lacked cytologic anaplasia and mitoses, making a diagnosis of tubulopapillary adenomas (Fig. 2).
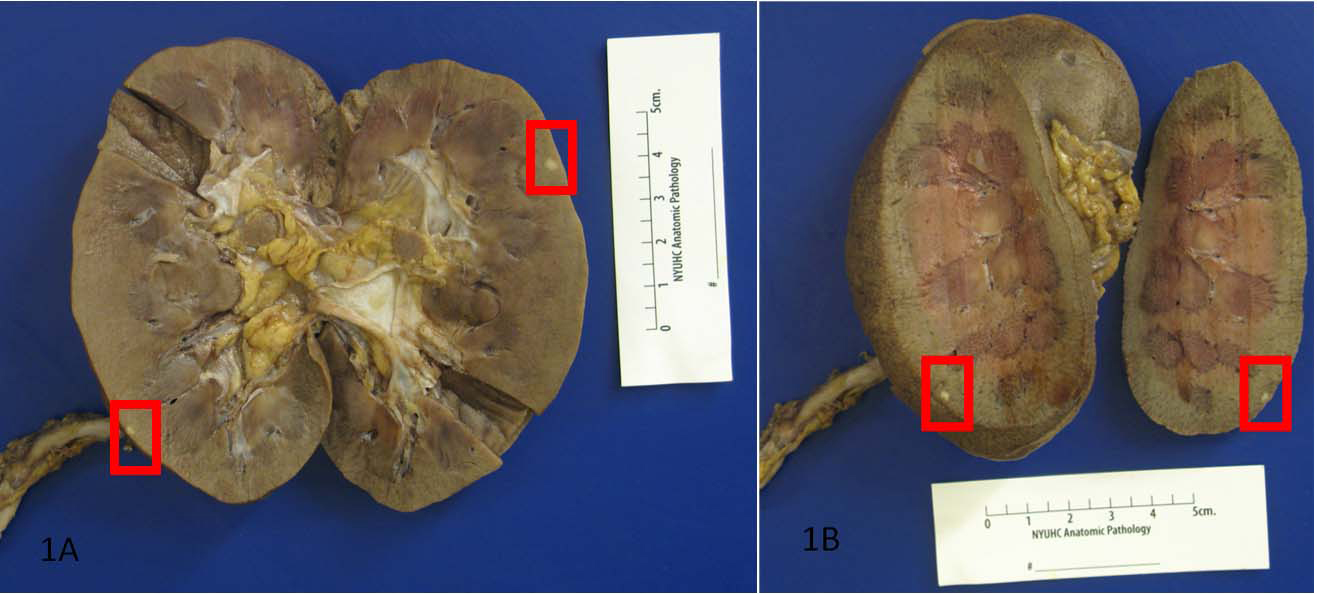 Click for large image | Figure 1. Autopsy revealed multiple well-circumscribed unencapsulated, pale yellow tumors in the subcapsular portions of both kidneys. |
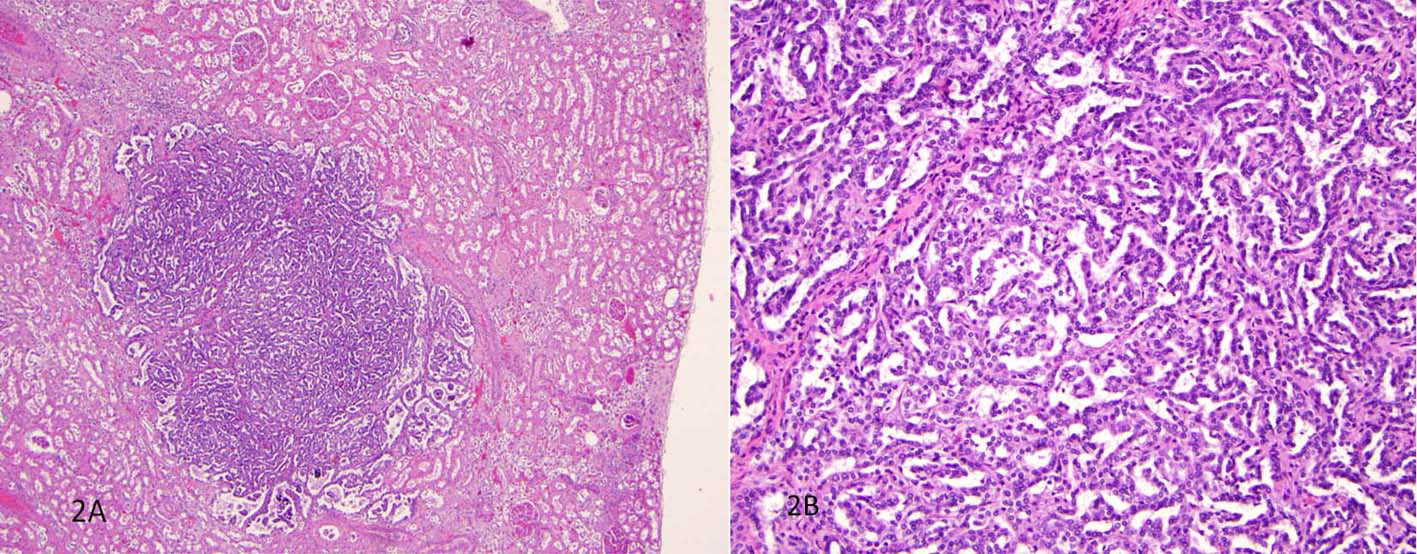 Click for large image | Figure 2. H&E sections revealed densely packed papillae or tubules lined by small, regular, cuboidal cells with rounded, uniform nuclei that lacked cytologic anaplasia and mitoses. |
| Discussion | ▴Top |
The mechanism of tumorgenesis in familial dysautonomia is unknown. A recent retrospective review indicates that two percent of patients with familial dysautonomia had a neoplasm [11]. All were detected prior to death between the ages of 7 to 49, which suggests that mutations in the IKBKAP gene may be predisposed to neoplasia at an early age. Therefore, considering that most of these patients succumb to the complications of their disease at a young age and that religious Jews often refuse to allow autopsy, it is likely that other patients with familial dysautonomia, like our own case, may also have died with unrecognized tumors which would increase the incidence of neoplasias even further. Notably, the father of this patient passed away at a young age of unknown cancer. It may be of significance since carriers are known to have reduced levels of the IKBKAP gene. In one study, a strong correlation was found between the IKBKAP mRNA levels of the 22 familial dysautonomia patients and their parents [12], suggesting that the efficiency of the splicing is controlled by genetic mechanisms.
In our case, we found multiple bilateral tubulopapillary adenomas, which are low-grade papillary lesions with striking histological and genetic similarities to papillary carcinomas and are distinguished from the latter on the basis of size under 0.5 cm [13]. These small renal cortical epithelial neoplasms may be found incidentally. However, bilateral multifocal lesions are uncommon, unless they develop as a part of von Hippel-Lindau syndrome [14]. Although the previous review of neoplasms in familial dysautonomia included some low-grade neoplastic lesions [11], with time these proliferative lesions may have evolved into malignancy if the decedents had a full life expectancy.
The relationship between our findings and the IKBKAP gene mutations is unclear. Cytogenetic and molecular techniques demonstrated balanced translocations involving chromosome 9p and 9q (q34) in both clear cell and papillary renal cell carcinoma, the malignant counterparts of the adenomas [15, 16]. Perhaps, the proximity to the causative gene may play a role in this association. Alternatively, by virtue of affecting the critical part of the transcriptional machinery, IKBKAP gene expression may be involved in crucial signaling cascades affecting expression of multiple secondary genes, the effects of which are yet to be discovered. Finally, defective repair of damaged DNA may provide yet another mechanism for accelerated tumorgenesis in patients with familial dysautonomia.
Conflict of Interest
The authors declare that they have no conflict of interest.
Funding
H.K. receives research support from the NIH (U54NS065736 [PI]), the FDA (FD-R-3731-01 [PI]), and the Dysautonomia Foundation, Inc.
| References | ▴Top |
- Slaugenhaupt SA, Blumenfeld A, Gill SP, Leyne M, Mull J, Cuajungco MP, Liebert CB,
et al . Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am J Hum Genet. 2001;68(3):598-605.
pubmed doi - Anderson SL, Coli R, Daly IW, Kichula EA, Rork MJ, Volpi SA, Ekstein J,
et al . Familial dysautonomia is caused by mutations of the IKAP gene. Am J Hum Genet. 2001;68(3):753-758.
pubmed doi - Norcliffe-Kaufmann L, Axelrod F, Kaufmann H. Afferent baroreflex failure in familial dysautonomia. Neurology. 2010;75(21):1904-1911.
pubmed doi - Johansen LD, Naumanen T, Knudsen A, Westerlund N, Gromova I, Junttila M, Nielsen C,
et al . IKAP localizes to membrane ruffles with filamin A and regulates actin cytoskeleton organization and cell migration. J Cell Sci. 2008;121(Pt 6):854-864.
pubmed doi - Otero G, Fellows J, Li Y, de Bizemont T, Dirac AM, Gustafsson CM, Erdjument-Bromage H,
et al . Elongator, a multisubunit component of a novel RNA polymerase II holoenzyme for transcriptional elongation. Mol Cell. 1999;3(1):109-118.
pubmed doi - Cheishvili D, Maayan C, Smith Y, Ast G, Razin A. IKAP/hELP1 deficiency in the cerebrum of familial dysautonomia patients results in down regulation of genes involved in oligodendrocyte differentiation and in myelination. Hum Mol Genet. 2007;16(17):2097-2104.
pubmed doi - Naumanen T, Johansen LD, Coffey ET, Kallunki T. Loss-of-function of IKAP/ELP1: could neuronal migration defect underlie familial dysautonomia? Cell Adh Migr. 2008;2(4):236-239.
pubmed - Close P, Hawkes N, Cornez I, Creppe C, Lambert CA, Rogister B, Siebenlist U,
et al . Transcription impairment and cell migration defects in elongator-depleted cells: implication for familial dysautonomia. Mol Cell. 2006;22(4):521-531.
pubmed doi - Chen YT, Hims MM, Shetty RS, Mull J, Liu L, Leyne M, Slaugenhaupt SA. Loss of mouse Ikbkap, a subunit of elongator, leads to transcriptional deficits and embryonic lethality that can be rescued by human IKBKAP. Mol Cell Biol. 2009;29(3):736-744.
pubmed doi - Brennan S, Lewis PD. Lack of hypersensitivity to ionising radiation in familial dysautonomia. Lancet. 1983;1(8325):657.
pubmed doi - Gold-von Simson G, Romanos-Sirakis E, Maayan C, Axelrod FB. Neoplasia in familial dysautonomia: a 20-year review in a young patient population. J Pediatr. 2009;155(6):934-936.
pubmed doi - Gold-von Simson G, Leyne M, Mull J, Rolnitzky LM, Goldberg JD, Berlin D, Axelrod FB,
et al . IKBKAP mRNA in peripheral blood leukocytes: a molecular marker of gene expression and splicing in familial dysautonomia. Pediatr Res. 2008;63(2):186-190.
pubmed doi - Storkel S, Eble JN, Adlakha K, Amin M, Blute ML, Bostwick DG, Darson M,
et al . Classification of renal cell carcinoma: Workgroup No. 1. Union Internationale Contre le Cancer (UICC) and the American Joint Committee on Cancer (AJCC). Cancer. 1997;80(5):987-989.
pubmed - Murphy WM, Grignon DJ and EJ Perlman. AFIP Atlas of Tumor Pathology: Tumors of the Kidney, Bladder and Related Urinary Structures 2004 (AFIP Atlas of Tumor Pathology 4th Series) Washington, DC: American Registry of Pathology.
- Sanjmyatav J, Schubert J, Junker K. Comparative study of renal cell carcinoma by CGH, multicolor-FISH and conventional cytogenic banding analysis. Oncol Rep. 2005;14(5):1183-1187.
pubmed - Shridhar V, Sun QC, Miller OJ, Kalemkerian GP, Petros J, Smith DI. Loss of heterozygosity on the long arm of human chromosome 7 in sporadic renal cell carcinomas. Oncogene. 1997;15(22):2727-2733.
pubmed doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.