

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website http://www.wjon.org |
Case Report
Volume 3, Number 3, June 2012, pages 127-133
Meningioangiomatosis Without Neurofibromatosis Type 2
Sara Marzia, Danilo De Paulisa, Alessandro Riccia, Graziano Taddeia, d, Soheila Raysi Dehcordia, Gino Colettib, Giuliano Masellia, Renato J. Galzioa, c
aDepartment of Neurosurgery, San Salvatore Hospital, L’Aquila, Italy
bDepartment of Pathology, San Salvatore Hospital, L’Aquila, Italy
cDepartment of Health Sciences, San Salvatore Hospital, L’Aquila, Italy
dCorresponding author: Graziano Taddei, Ospedale S. Salvatore, via Vetoio 1, 67100 L’Aquila, Italy
Manuscript accepted for publication February 14, 2012
Short title: Meningioangiomatosis not Associated With NF-2
doi: https://doi.org/10.4021/wjon470w
| Abstract | ▴Top |
Meningioangiomatosis (MA) is a rare, benign hamartomatous lesion found in cerebral cortex and leptomeninges. It occurs mostly in 5 - 15 year old children in form isolated or diffuse; the diffuse form may be associated with neurofibromatosis type 2 (NF2). The sporadic type in the adults is less common.The patient was a 37 year-old man with a long history of frontal headache. In suspected sinusitis, the patient underwent cerebral MRI that showed hypointense lesion in the right frontal lobe with heterogeneous contrast enhancement after gadolinium administration. There were no stigmata or family history of neurofibromatosis. A right pterional approach with a supraorbital craniotomy was performed. The lesion was removed with complete remission of the headache in the postoperative time. MA enters into differential diagnosis with several other diseases and a correct diagnosis is mandatory. The total surgical removal is the treatment of choice, and the prognosis after surgery is usually excellent for the absence of recurrence in sporadic cases.
Keywords: Meningioangiomatosis; Cerebral hamartoma; Neurofibromatosis type 2
| Introduction | ▴Top |
Meningioangiomatosis (MA) is a rare, benign hamartomatous lesion found in cerebral cortex and leptomeninges [1]. It is characterized by intracortical proliferation of meningothelial cells, microvasculature and fibroblasts that may mimic a neoplasm clinically and radiologically [2] MA may occur mostly in 5 - 15 year old children and may be isolated or diffuse; the diffuse form may be associated with neurofibromatosis type 2 (NF2) [3]. Sporadic MA usually presents seizures and persistent headaches, while NF2-associated forms are often asymptomatic and diagnosed only at autopsy [4]. The authors describe a case of sporadic form of MA unassociated with NF2 reviewing the pertinent literature.
| Case Report | ▴Top |
A 37-year-old man was admitted to our hospital complaining of frequent episodes of headache for the past several months. He was followed by the otolaryngologist for sinusitis, but for the persistence of the headache, computed tomography (CT) and magnetic resonance (MR) of the brain were performed. CT scans showed a right frontal calcified lesion e (Fig. 1) and marked enhancement post contrast and MR revealed hypointense lesion in the right frontal lobe with heterogeneous contrast enhancement after Gadolinium administration (Fig. 2). On admission to our department, general, physical and neurological examination found no abnormalities. X-ray scan of the skull revealed an extended geographical area of osteolysis in charge of the frontal squama in the right supraorbital region with involvement of the roof of the ipsilateral orbit and hypoplasia of frontal sinus. A brain CT and MR were repeated and showed no changes beyond those already performed. There were no stigmata or family history of neurofibromatosis.
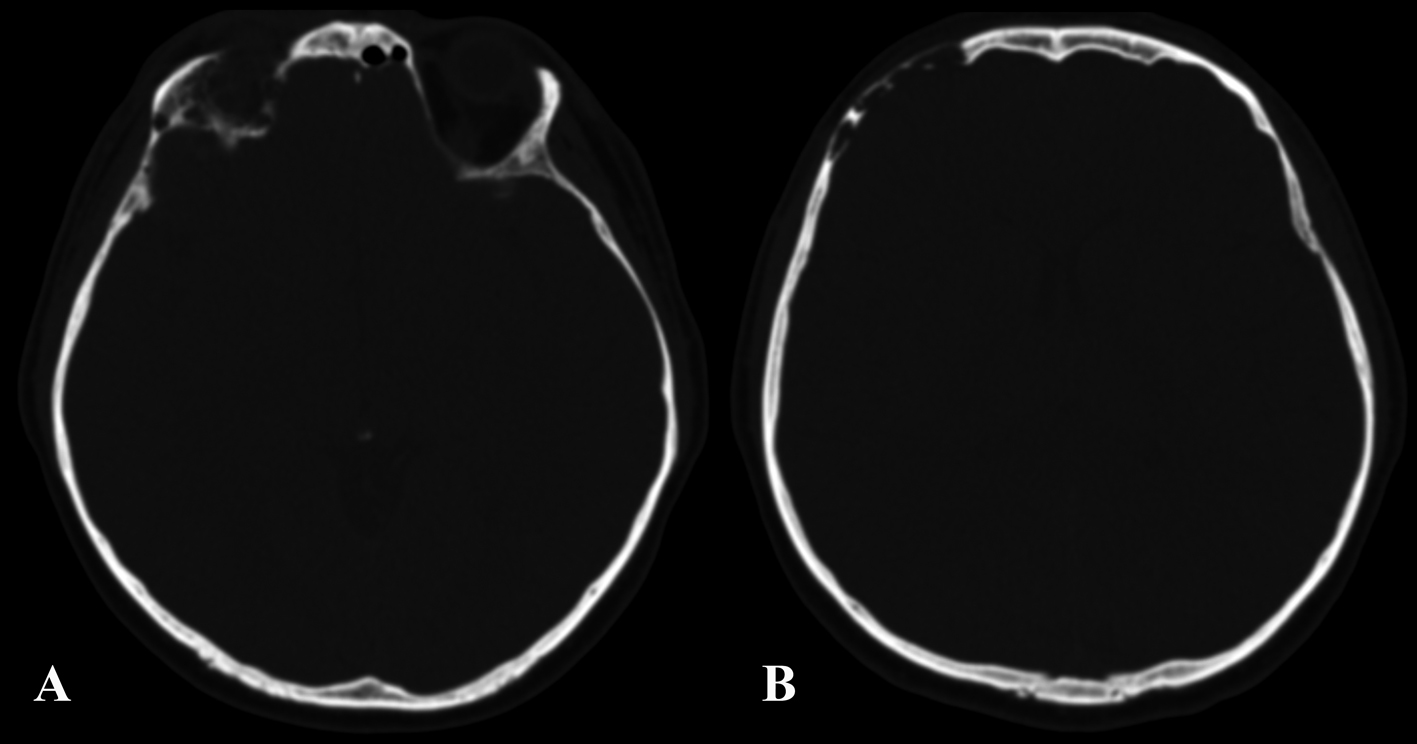 Click for large image | Figure 1. Pre-operative CT showing the involvement of the right orbital roof (A) and the right frontal bone (B) by the lesion. |
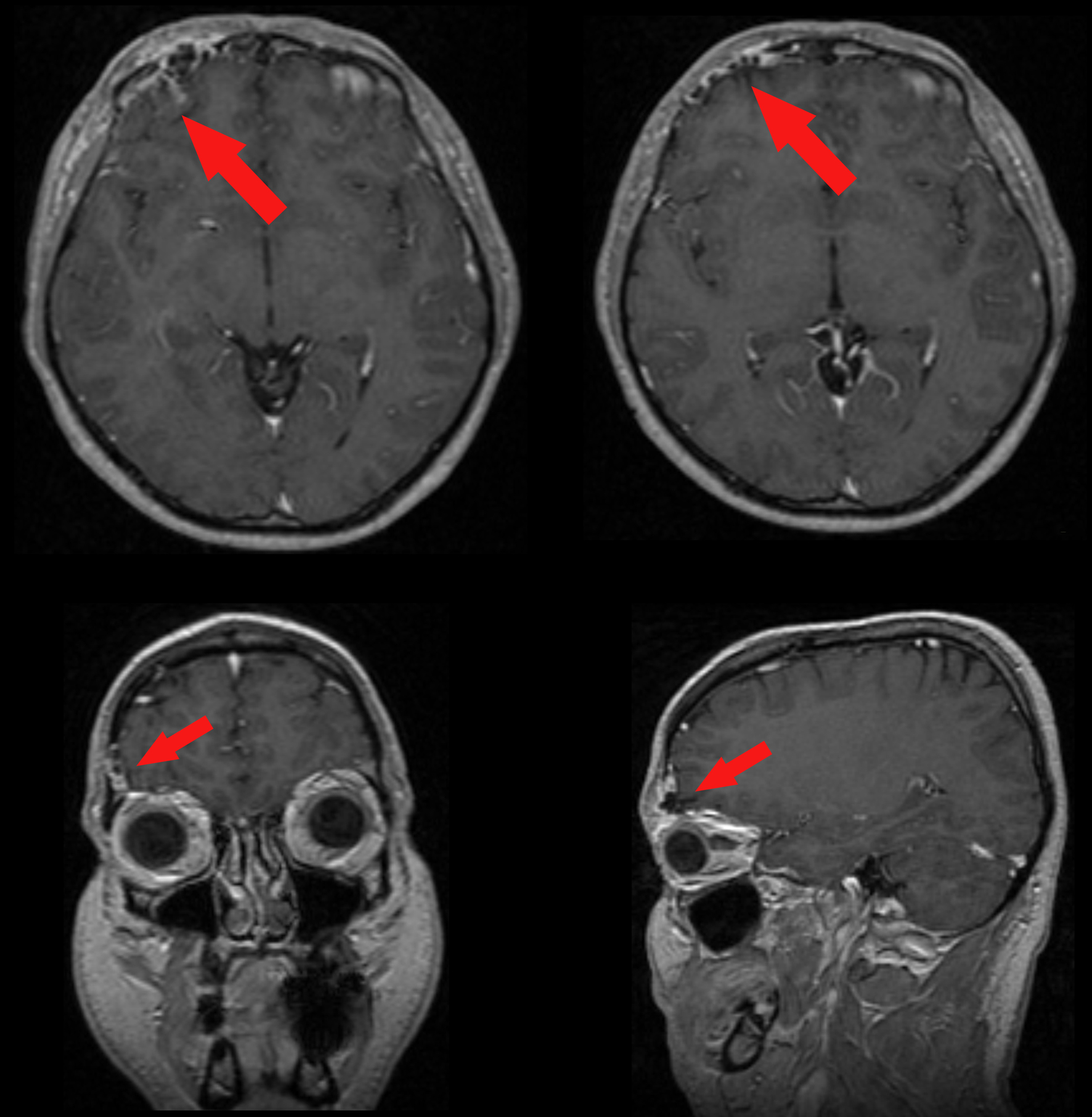 Click for large image | Figure 2. Pre-operative enhanced MR showing the meningioangiomatosis (red arrows) in the 3-plains projection. |
A right pterional approach was performed. The bone presented a gap full of blood sized 3 × 2.5 cm. The craniotomy was performed round this gap with a margin of 1 cm. The roof of the orbit ipsilateral was involved by the fibrous part of the lesion, which was not well demarcated from the surrounding soft brain parenchyma. We performed a progressive vascular deafferentation and disconnection of the lesion from the parenchyma to its anteromedial segment that appeared in proximity to the base of the superior sagittal sinus through the vein of Sperino. Dura appeared thickened, well vascularized and its dissection from the parenchyma was difficult. At the end, the alterated dural surface was replaced by a patch of galea and the bone reconstruction was obtained using his bony flap.
Histological examination showed areas of collagen "dura-like" (clearly visible after staining with Masson's trichrome) accompanied by areas of bone metaplasia and presence of cerebral gray matter with normal architecture. There was a rich vascularization with vessels often with wall hyalinization, perivascular fibroblastic spindle cells and rare psammoma bodies. Coexisting areas, including extensive benign MA proliferation (EMA +), especially in the perivascular site, were present (Fig. 3).
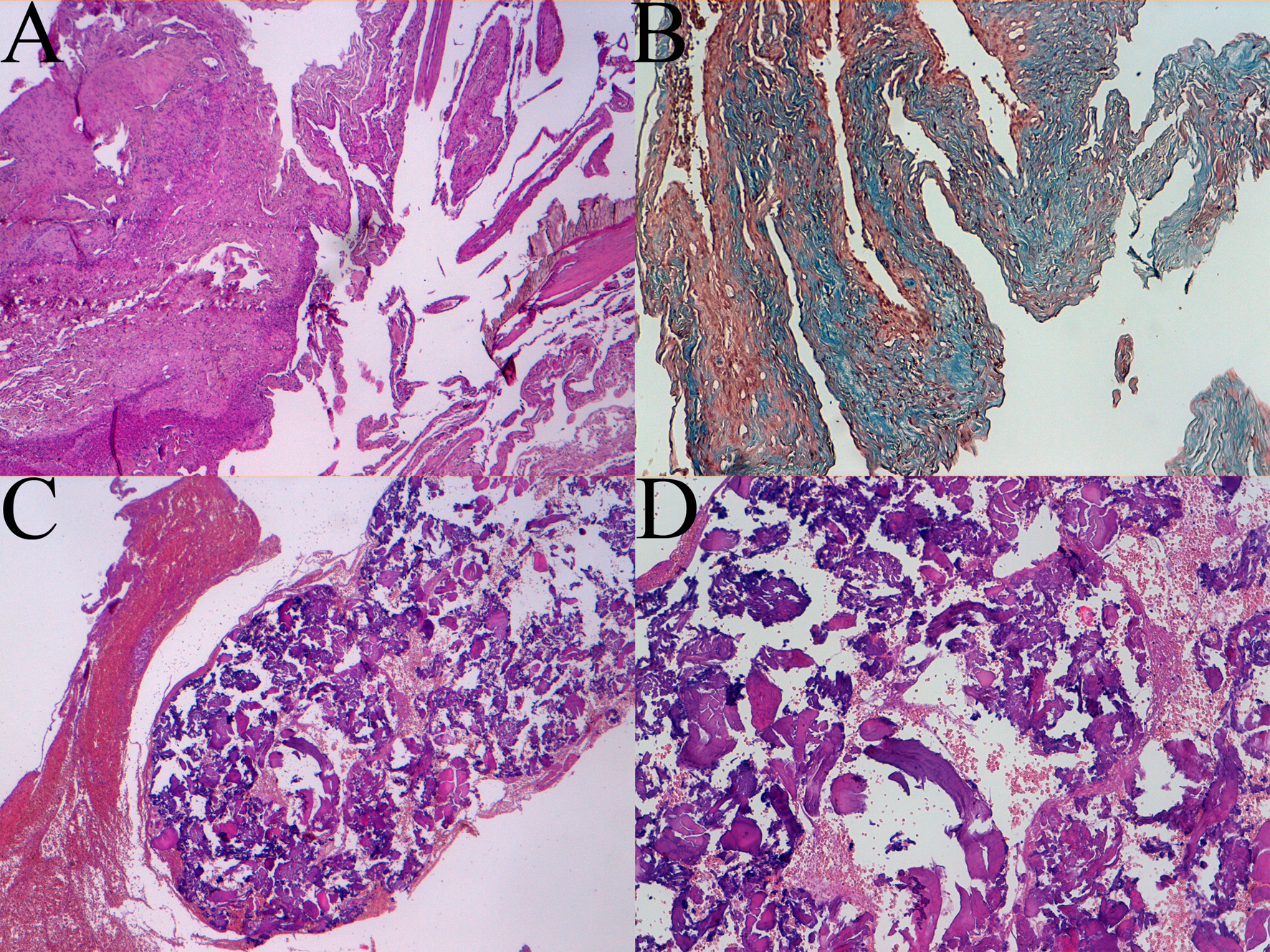 Click for large image | Figure 3. Histologic patterns of meningioangiomatosis: A sclerotic areas (4 × magnification), B trichrome coloration view (10 × magnification), C and D bone metaplasia at 4 × and 10 × magnification respectively. |
The patient’s postoperative course was uneventful. At 6 months follow up, the patient reported no more episodes of headache (Fig. 4, 5).
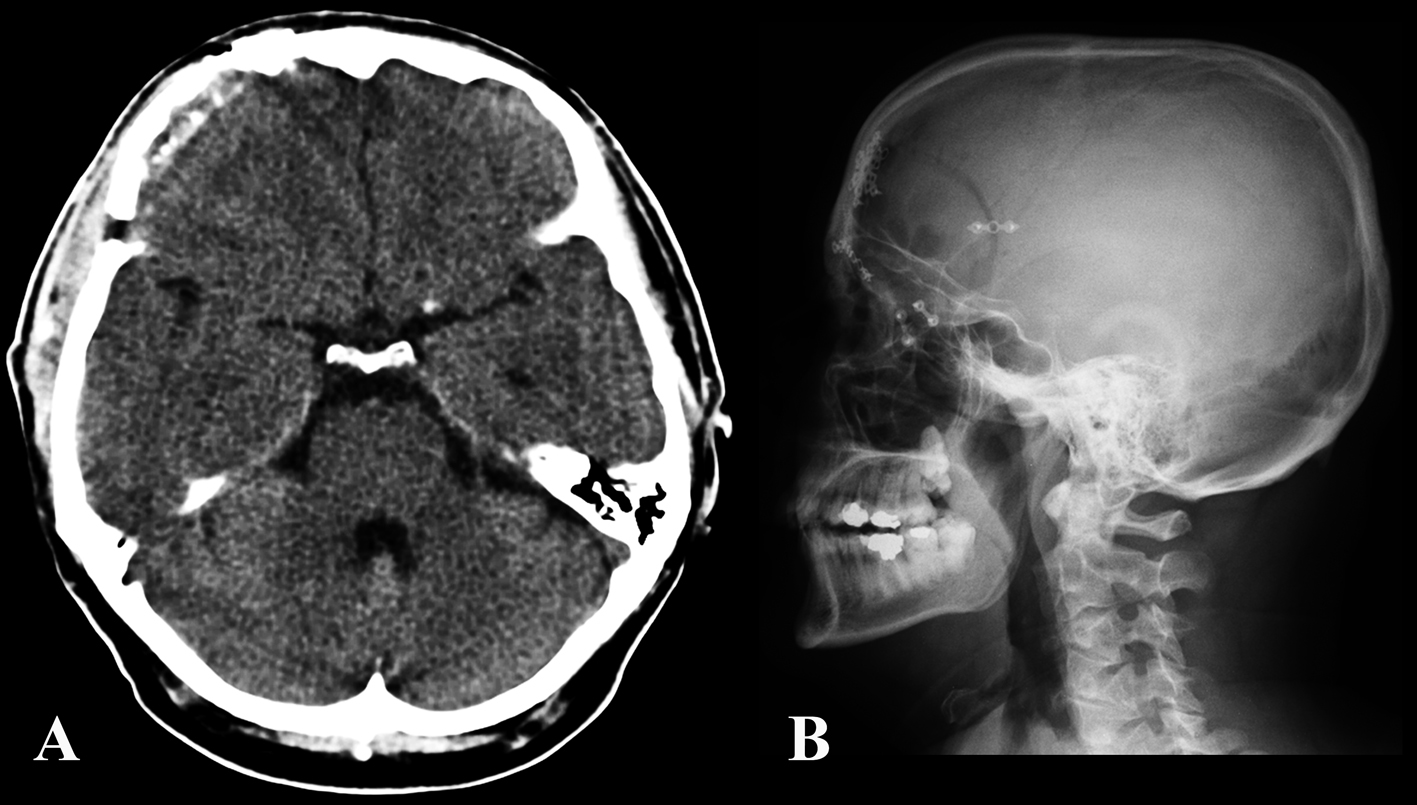 Click for large image | Figure 4. Post-operative CT (A) and skull x-plain film (B) showing the craniotomy and bony reconstruction. |
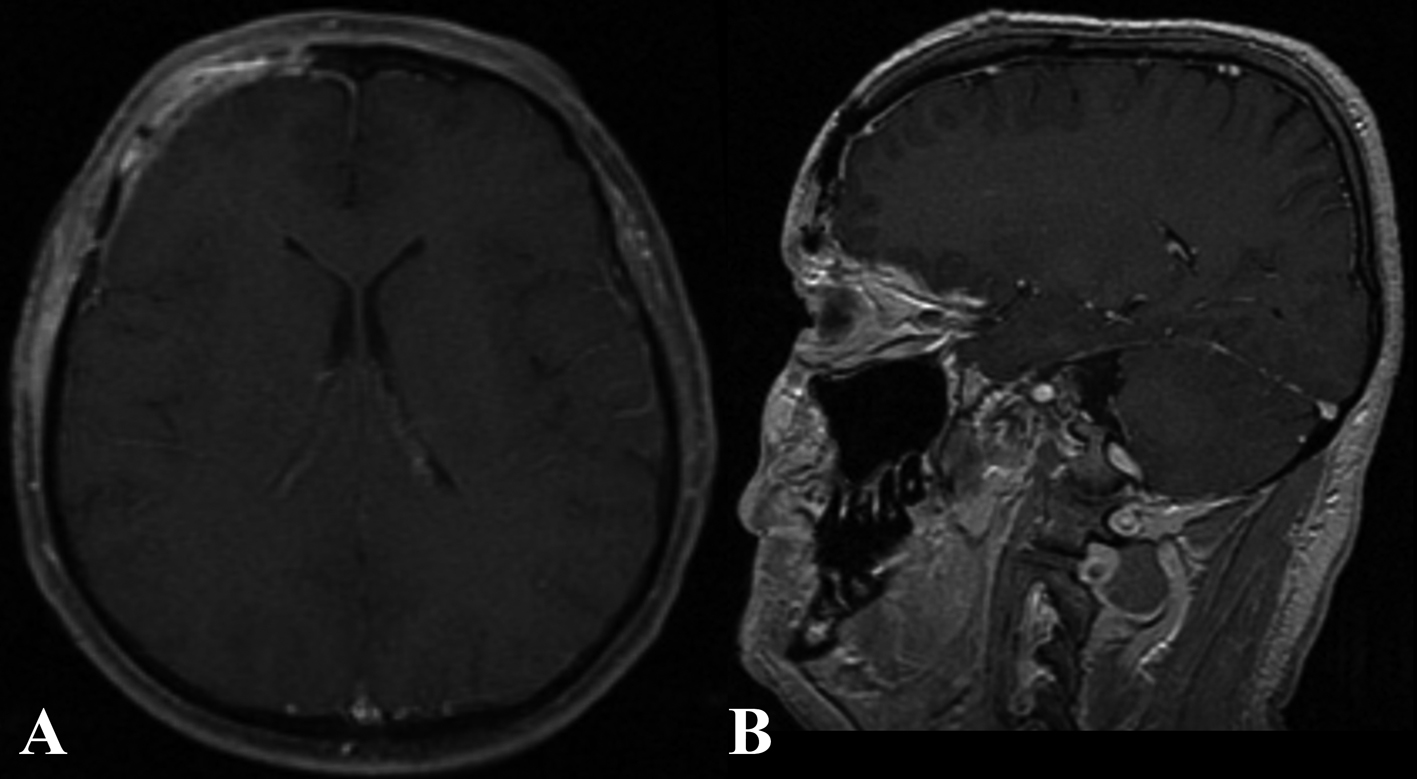 Click for large image | Figure 5. Six months post-operative enhanced MR in the axial (A) and sagittal (B) plain. |
| Discussion | ▴Top |
MA is a rare condition in the adult patients. Only 18 reports with a total of 23 adult patients have been described in literature [1, 2, 4-19]. The average age of the patients reported is 35 years (range 18 - 71) with a relative male prevalence (14:9). The predominant presenting symptoms are seizures (18 cases) and headache (3 cases) but in 2 cases drowsiness and weakness are respectively the initial symptoms. The association with NF2 was described in only 1 case (Table 1).
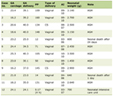 Click to view | Table 1. Reviewing of the Literature of Meningioangiomatosis in the Adult Patients |
Generally, MA occurs both in sporadic and syndromic forms, the latter with NF2 [2]. Most cases are asymptomatic; the symptomatic patients tend to present during childhood and early adulthood, usually with seizures. The symptoms are correlated with features of increased intracranial pressure [16]. The tumor may be located in frontal, temporal or parietal cortex with some reports in the third ventricle, cingulate gyrus, pulvinar and brainstem [2, 20].
It was first described by Bassoe and Nuzum in 1915 [21] as an incidental autopsy in a 15-year old boy originally described in association with von Recklinghausen’s disease but it was named by Worster-Drought in 1937 [22]. It is a rare benign disorder characterized by the histological hallmarks of meningiomas and angiomas [4]. In the past, the pathogenesis and histopathological pleomorphism has created diagnostic difficulties [18]. Formerly, MA was considered as a hamartomatous lesion with degenerative changes [4, 21]. Kasanticul and Brown [23] suggested a direct invasion of brain tissue from a leptomeningeal meningioma. Other theories include cortical vascular malformation [24, 25], or the initial development of angiomatous tissue with meningioangiomatous components arising secondarily from perivascular elements [16]. However, even though some studies revealed the loss of 22q12 (NF2 gene) in cases of MA and MA- associated meningioma indicating the possibility of a neoplastic nature [4, 24, 26, 27], a large genetic series demonstrated that there was no NF2 gene mutation either in sporadic or NF2-associated MA in according with the hypothesis of a hamartomatous nature [27, 28].
Generally, solitary MA lesions measuring up to 5 cm are accompanied by meningeal thickening and calcification producing a tan-yellow and gritty tissue [29]. The tumor is usually composed of two components: a leptomeningeal meningothelial proliferation and a meningo-vascular proliferation. The intraparenchymal proliferation of small vessels is accompanied by concentric perivascular proliferation of spindle-shaped meningeal cells similar to those found in a meningioma [20]. The overlying leptomeningeal components of proliferating meningothelial cells can produce marked degenerative reactions such as gliosis, perivascular connective tissue proliferation, dysplastic neurons, white matter cysts and large-vessel hyalinization, calcification, psammoma bodies, fibrocartilage and bone formations [18, 20]. In many cases, proliferating perivascular cells infiltrate the cortex in association with marked cellularity and reactive gliosis [18]. For these reasons, MA can be divided into 3 types: predominantly cellular type, vascular type, and fibrocalcifying type [29]. On the contrary, the presence of neurofibrillary tangles with or without amyloid plaques and granulo-vacuolar degeneration represent a reactive phenomenon rather than an intrinsic lesion component [18].
Although the pre-operative evaluation results difficult because of diagnostic tools lack specificity can be found elements for a correct diagnosis. On the CT scan, these lesions can underline the presence of calcification with surrounding low-density edema. On MRI, they exhibited central low- or mixed-signal intensity on T1-weighted images and a surrounding high-signal edema on T2-weighted images. The enhancement after administration of Gadolinium was seen not in all cases, varying from slight to strong heterogeneous enhancement or even no enhancement at all [14, 18, 30, 31]. It seemed that enhancement patterns had correlation with the proliferating leptomeningeal microvessels [31]. However, MRI can be useful to demonstrate the cortical location of MA, associated with gyriform hyperintensity on FLAIR sequence, which correlated with proliferating microvessels with perivascular cuffs of spindle-cell proliferation within the cortex [31].
The electrophysiological evaluation of MA with and without seizures is complex to analyze with a wide array of findings. Generally, the lesion shows a range of electrophysiological abnormalities. It can encompass circumscribed background slowing and epileptogenicity, multifocal extralesional spikes and sharp waves, and generalized spike-waves without definitive secondary bilateral synchrony or shows an independent and synchronous epileptiform discharges on the surface and within (depth) the MA but not in the adjacent neocortex. However, it can be also electrographically quiescent [18, 19].
Total surgical removal is the treatment of choice for MA. Normally, in these cases the lesion is not growing again and the symptoms usually disappear as occurred in our experience [15, 17, 32]. However in literature seizure-free rates after resection are variably between 43 - 68% of the cases and almost 70 - 80% of the patients continued to required antiepileptic drugs [32, 33].
These results probably arise from a partial surgical resection. In fact, the lesion can extend into the perivascular space but spares the white matter with cortical neurons and rare neurofibrillary tangles entrapped within the lesion [33, 34]. However, the absence of pleomorphism, mitoses, necrosis, cortical invasions or calcifications, that are often associated with MA, cannot allow the complete surgical resection.
In conclusion, MA enters into differential diagnosis with several other diseases such as oligodendroglioma, granulomatous meningitis, meningioma, vascular lesions such as Sturge-Weber syndrome and arteriovenous malformation and parasitic diseases. A correct diagnosis is mandatory because it is a benign lesion that does not become malignant. The total surgical removal is the treatment of choice, and the prognosis after surgery is usually excellent for the absence of recurrence in sporadic cases.
Conflict of Interest
The authors do not have any conflict of interest or financial arrangement.
| References | ▴Top |
- Park MS, Suh DC, Choi WS, Lee SY, Kang GH. Multifocal meningioangiomatosis: a report of two cases. AJNR Am J Neuroradiol. 1999;20(4):677-680.
pubmed - Savargaonkar P, Chen S, Bhuiya T, Valderrama E, Bloom T, Farmer PM. Meningioangiomatosis: report of three cases and review of the literature. Ann Clin Lab Sci. 2003;33(1):115-118.
pubmed - Kim SH, Yoon SH, Kim JH. A case of infantile meningioangiomatosis with a separate cyst. J Korean Neurosurg Soc. 2009;46(3):252-256.
pubmed doi - Chen YY, Tiang XY, Li Z, Luo BN, Huang Q. Sporadic meningioangiomatosis-associated atypical meningioma mimicking parenchymal invasion of brain: a case report and review of the literature. Diagn Pathol. 2010;5:39.
pubmed - Aizpuru RN, Quencer RM, Norenberg M, Altman N, Smirniotopoulos J. Meningioangiomatosis: clinical, radiologic, and histopathologic correlation. Radiology. 1991;179(3):819-821.
pubmed - Chakrabarty A, Franks AJ. Meningioangiomatosis: a case report and review of the literature. Br J Neurosurg. 1999;13(2):167-173.
pubmed doi - Giangaspero F, Guiducci A, Lenz FA, Mastronardi L, Burger PC. Meningioma with meningioangiomatosis: a condition mimicking invasive meningiomas in children and young adults: report of two cases and review of the literature. Am J Surg Pathol. 1999;23(8):872-875.
pubmed doi - Halper J, Scheithauer BW, Okazaki H, Laws ER
Jr . Meningio-angiomatosis: a report of six cases with special reference to the occurrence of neurofibrillary tangles. J Neuropathol Exp Neurol. 1986;45(4):426-446.
pubmed doi - Huson SM, Harper PS, Compston DA. Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain. 1988;111(Pt 6):1355-1381.
pubmed - Kunishio K, Yamamoto Y, Sunami N, Satoh T, Asari S, Yoshino T, Ohtuki Y. Histopathologic investigation of a case of meningioangiomatosis not associated with von Recklinghausen's disease. Surg Neurol. 1987;27(6):575-579.
pubmed doi - Kuzniecky R, Melanson D, Robitaille Y, Olivier A. Magnetic resonance imaging of meningio-angiomatosis. Can J Neurol Sci. 1988;15(2):161-164.
pubmed - Liu SS, Johnson PC, Sonntag VK. Meningioangiomatosis: a case report. Surg Neurol. 1989;31(5):376-380.
pubmed doi - Matias-Guiu X, Molet J, Ferrer I, Prat J. Meningo-angiomatosis: a case report. Br J Neurosurg. 1988;2(1):97-100.
pubmed doi - Mut M, Soylemezoglu F, Firat MM, Palaoglu S. Intraparenchymal meningioma originating from underlying meningioangiomatosis. Case report and review of the literature. J Neurosurg. 2000;92(4):706-710.
pubmed doi - Prayson RA. Meningioangiomatosis. A clinicopathologic study including MIB1 immunoreactivity. Arch Pathol Lab Med. 1995;119(11):1061-1064.
pubmed - Sakaki S, Nakagawa K, Nakamura K, Takeda S. Meningioangiomatosis not associated with von Recklinghausen's disease. Neurosurgery. 1987;20(5):797-801.
pubmed doi - Tacconi L, Thom M, Symon L. Cerebral meningioangiomatosis: case report. Surg Neurol. 1997;48(3):255-260.
pubmed doi - Whiting DM, Awad IA, Miles J, Chou SS, Luders H. Intractable complex partial seizures associated with occult temporal lobe encephalocele and meningoangiomatosis: a case report. Surg Neurol. 1990;34(5):318-322.
pubmed doi - Wiebe S, Munoz DG, Smith S, Lee DH. Meningioangiomatosis. A comprehensive analysis of clinical and laboratory features. Brain. 1999;122(Pt 4):709-726.
pubmed - Kollias SS, Crone KR, Ball WS
Jr , Prenger EC, Ballard ET. Meningioangiomatosis of the brain stem. Case report. J Neurosurg. 1994;80(4):732-735.
pubmed doi - Bassoe P, Nuzum F. Report of a case of central and peripheral neurofibromatosis. J Nerv Ment Dis. 1915;42:785-796.
- Worster-Drought C, Dickson WEC, McMenemey WH. Multiple meningeal and perineural tumors with analogous changes in glia and ependyma (neurofibroblastomatosis). Barin. 1937;60:85-117.
- Kasantikul V, Brown WJ. Meningioangiomatosis in the absence of von Recklinghausen's disease. Surg Neurol. 1981;15(1):71-75.
pubmed doi - Greene JF
Jr , Fitzwater JE, Burgess J. Arterial lesions associated with neurofibromatosis. Am J Clin Pathol. 1974;62(4):481-487.
pubmed - Malecha MJ, Rubin R. Aneurysms of the carotid arteries associated with von Recklinghausen's neurofibromatosis. Pathol Res Pract. 1992;188(1-2):145-147.
pubmed - Sinkre P, Perry A, Cai D, Raghavan R, Watson M, Wilson K, Barton Rogers B. Deletion of the NF2 region in both meningioma and juxtaposed meningioangiomatosis: case report supporting a neoplastic relationship. Pediatr Dev Pathol. 2001;4(6):568-572.
pubmed doi - Stemmer-Rachamimov AO, Horgan MA, Taratuto AL, Munoz DG, Smith TW, Frosch MP, Louis DN. Meningioangiomatosis is associated with neurofibromatosis 2 but not with somatic alterations of the NF2 gene. J Neuropathol Exp Neurol. 1997;56(5):485-489.
pubmed doi - Perry A, Kurtkaya-Yapicier O, Scheithauer BW, Robinson S, Prayson RA, Kleinschmidt-DeMasters BK, Stemmer-Rachamimov AO,
et al . Insights into meningioangiomatosis with and without meningioma: a clinicopathologic and genetic series of 24 cases with review of the literature. Brain Pathol. 2005;15(1):55-65.
pubmed doi - Wang Y, Gao X, Yao ZW, Chen H, Zhu JJ, Wang SX, Gao MS,
et al . Histopathological study of five cases with sporadic meningioangiomatosis. Neuropathology. 2006;26(3):249-256.
pubmed doi - Tien RD, Osumi A, Oakes JW, Madden JF, Burger PC. Meningioangiomatosis: CT and MR findings. J Comput Assist Tomogr. 1992;16(3):361-365.
pubmed doi - Yao Z, Wang Y, Zee C, Feng X, Sun H. Computed tomography and magnetic resonance appearance of sporadic meningioangiomatosis correlated with pathological findings. J Comput Assist Tomogr. 2009;33(5):799-804.
pubmed doi - Arcos A, Serramito R, Santin JM, Prieto A, Gelabert M, Rodriguez-Osorio X, Reyes R. Meningioangiomatosis: clinical-radiological features and surgical outcome. Neurocirugia (Astur). 2010;21(6):461-466.
pubmed doi - Jallo GI, Kothbauer K, Mehta V, Abbott R, Epstein F. Meningioangiomatosis without neurofibromatosis: a clinical analysis. J Neurosurg. 2005;103(4 Suppl):319-324.
pubmed - Wixom C, Chadwick AE, Krous HF. Sudden, unexpected death associated with meningioangiomatosis: case report. Pediatr Dev Pathol. 2005;8(2):240-244.
pubmed doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.