

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Review
Volume 13, Number 1, February 2022, pages 8-19

Tumor Dormancy: Biologic and Therapeutic Implications
Vanshika Balayana, Achuta Kumar Guddatia, b
aDivision of Hematology/Oncology, Georgia Cancer Center, Augusta University, Augusta, GA 30912, USA
bCorresponding Author: Achuta Kumar Guddati, Division of Hematology/Oncology, Georgia Cancer Center, Augusta University, Augusta, GA 30912, USA
Manuscript submitted September 30, 2021, accepted November 24, 2021, published online February 8, 2022
Short title: Tumor Dormancy Mechanisms
doi: https://doi.org/10.14740/wjon1419
| Abstract | ▴Top |
Metastatic cancer can arise years after treatment of the primary tumor because residual tumor cells can enter dormancy and evade elimination by anti-neoplastic therapies. The mechanisms underlying this phenomenon have been investigated and a number of hypotheses have been proposed. Tumor mass dormancy involves a balance between apoptotic and proliferative cells, keeping a micrometastatic lesion constant in size. This induces a need for blood supply which involves angiogenic dormancy. Cellular dormancy is also considered a mechanism of dormancy, where dormancy is induced due to cells entering a quiescent, reversible, growth-arrested state. In addition to all of these mechanisms, important changes in the tumor microenvironment, including the extracellular matrix, the oxygenation levels of the environment, and endoplasmic reticulum stress, are involved in inducing and maintaining tumor dormancy. Since dormant tumors are commonly known to be resistant to chemotherapy, gaining more knowledge of the mechanism of dormant tumor cells is of importance, as it can lead to the development of future treatment strategies.
Keywords: Tumor dormancy; Niche; Metastasis; Relapse; Recurrence
| Introduction | ▴Top |
In many types of tumors, the metastatic disease can occur years after resection of the tumor. Tumor relapse after a prolonged period can be explained by the survival of disseminated tumor cells (DTCs) in a dormant state [1]. These cells can disseminate early and reside in niches in distant organs before reactivating to cause a relapse of disease. This process of tumor dormancy has been seen in many common cancers, including colon cancer, kidney cancer, breast cancer, lung cancer, prostate cancer, melanoma, multiple myeloma, and leukemia [2]. The concept of tumor recurrence as first noted as by Celsus - a Roman physician of 25 BC to 50 AD era, has since developed as we have learned more. The period between the detection of the primary tumor and its metastatic relapse is referred to as tumor dormancy. In 1954, Geoffrey Hadfield laid the foundation for the concept of tumor cell dormancy and defined the term dormancy cancer cells as “non-proliferating cancer cells that have undergone G0/G1 phase cell cycle arrest” [3]. Tumor dormancy results from mechanisms that inhibit the further expansion of a dividing tumor cell population while also causing tumor cell growth arrest and putting the cells into a state of quiescence. This quiescent state is partially attributed to the up-regulation of p21 and p27, among other cell cycle inhibitors [1].
It has been postulated that cancer cells proliferate to form a micrometastatic lesion that does not expand beyond a certain size, likely caused by a balance between cell proliferation and apoptosis that is regulated by pro- and anti-angiogenic factors produced by the tumor microenvironment (TME). This phenomenon is called angiogenic dormancy. Any disruption in this balance could cause a transient angiogenic burst, which could interrupt the tumor dormancy period [4]. Tumor dormancy can also be maintained by the immune system, which does not eliminate the malignant tumor growth due to immune evasion, in a process called immunologic dormancy [1]. The third subdivision of tumor dormancy is cellular dormancy, where extrinsic and intrinsic mechanisms form a balance to allow for the formation of a quiescent state for tumor cells. This quiescent, dormant phase of tumors offers a promising therapeutic window for cancer treatment. The dormant stage can either present as the earliest stage in tumor development or as a stage in micro-metastases and minimal residual disease left after treatment and surgical resection of primary tumors [5]. It is also important to note that dormant tumor cells left after primary tumor removal and treatment are usually known to be refractory to chemotherapy [6]. While these disseminated dormant tumor cells may be minuscule, they can undergo a switch into a proliferative phase, allowing for the appearance of a fast-growing, clinically apparent tumor that is now resistant to chemotherapy. Dormancy may arise from a single cell that rests in a long-term dormant state, or that small groups of cells exist as micrometastases in equilibrium, resulting in no net size change [7]. This review goes over the currently known mechanisms of tumor dormancy induction, maintenance, and escape.
Table 1 [5, 8-41] provides a user-friendly interface that summarizes publications that can be accessed to gain further understanding of tumor dormancy mechanisms that are also cited in the discussion.
 Click to view | Table 1. Some Major Studies With Tumor Dormancy Discoveries |
| Discussion | ▴Top |
Difference between dormant cancer cells and cancer stem cells
A confounding factor in the study of tumor dormancy exists due to confusion between dormant cancer cells and cancer stem cells, and it is important to delineate a difference between the two before presenting a perspective on dormancy. Cancer stem cells were originally described in 1988 as a “mixture of malignant stem cells with marked capacity for proliferation and limited capacity for differentiation” [42, 43]. The existence of cancer stem cells has since been proven and seen in acute myeloid leukemia (AML), as well as breast, brain, colon, ovarian, and pancreatic cancers [44-46]. Both cancer stem cells and dormant cancer cells are considered to be drug-resistant and responsible for disease relapse, but there are a few key differences. Cancer stem cells are shown to slow down in the cell cycle, rather than complete cell cycle arrest [46]. Additionally, cancer stem cells undergo self-renewal, likely by asymmetric cell division, while active and dormant cancer cells are at the same stage of cell differentiation and are shown to switch between each other as needed [8, 42]. Dormant cells have a different gene expression profile than that of parental cells, where the regulation of certain genes is a critical point in the onset and maintenance of dormancy. For example, the thrombospondin-1 (TSP-1) and epidermal growth factor receptor (EGFR) are regulators of glioblastoma dormancy [47]. Dormant cancer cells exemplify niche dependence, cell cycle arrest, drug resistance, immune evasion, metastatic relapse, and reversibility [48]. It can even be considered that metastatic relapse occurs as a part of the progression of the dormant cancer cell life cycle.
Cancer can recur years after surgical resection in many types of cancers, especially breast and prostate cancer [49]. Human epidermal growth factor receptor 2 (HER2)-positive breast cancer and triple-negative breast cancer has a particularly high proliferation activity and tends to have a shorter mean time to distance recurrence than other subtypes of breast cancer [50]. In comparison, estrogen receptor (ER)-positive breast cancer has been shown to have cells that can remain dormant and survive for more than 5 years after primary tumor resection, requiring prolonged treatment [51, 52]. In metastatic breast cancer cells in the bone marrow, tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) have been shown to help remodel the bone microenvironment to cue dormant breast cancer cells out of dormancy and into proliferation [53]. It was also shown that these metastatic breast cancer cells were cued into dormancy by TSP-1 production by the nearby microvasculature [9].
Prostate cancer studies have shown several instances with recurrences occurring with elevated serum prostate-specific antigen (PSA) levels without any apparent recurrence of the tumor. This phenomenon is called PSA recurrence, and the recurrence of clinically evident tumors occurs on average 8 years after the PSA recurrence [54]. Prostate cancer cell lines were shown to become dormant when cocultured with a pre-osteoblastic cell line through the interaction between transforming growth factor beta-2 (TGF-β2) and the Axl ligand that is secreted by osteoblasts.
Role of the niche
The first stage of the dormant cancer cell life cycle requires that these cells find the right niche in which they can normally survive. In multiple myeloma, bone lining cells were shown to also be an endosteal niche for myeloma cells as in vivo studies showed that osteoclasts forced the myeloma cells out of dormancy and into a proliferative stage. These osteoclasts served as a switch, where dormancy was switched “on” by engagement with bone lining cells and switched “off” by osteoclast remodeling of the endosteal niche [8]. It is also likely that similar niches also exist for dormant cancer cells in other organs. Prostate and luminal breast cancers are known to prefer bone while triple-negative breast cancers have a predilection for visceral organs. These niche environments likely serve a series of different functions. The endosteal niche for myeloma cells works to support hematopoietic stem cells (HSCs) but can have their functions taken over by DTCs, leading to functional overlap between the DTCs and HSCs [10]. This makes studying the niche more difficult. For example, multiple myeloma patients were studied where HSCs were mobilized with granulocyte colony-stimulating factor (G-CSF) to increase the numbers of mobile monoclonal plasma cells [11]. However, adding CXC-chemokine receptor 4 (CXCR4) antagonist, which blocks binding of CXC-chemokine ligand 12 (CXCL12), to G-CSF did not change the rate of cancer cell immobilization in multiple myeloma patients [12]. CXCL12 has been suggested to regulate most aspects of HSC maintenance, proliferation, localization, and self-renewal, which exists in cells that make up the niche within which these dormant cells reside [13]. CXCL12 and the αvβ3 integrin secretion allow binding to bone matrix proteins to sustain these niches for prolonged periods [14]. Imaging has shown that multiple myeloma cells travel through the vascular bed without arresting in bone, demonstrating the increasing likelihood that these cells circulate until they find the right niche [8]. For breast and prostate cancer, the preference for an endosteal niche has been linked to CXCR4 expression as well [15, 16]. A study showed that breast cancer cells demonstrated upregulated genes that helped them gain entry into E-selectin+CXCL12+ perivascular niches in the bone marrow [17]. An immunodeficient mouse model showed human prostate cancer cells in competition with mouse HSCs for binding to annexin A2 of osteoblastic cells [18]. Both HSCs and prostate cancer cells co-localized with runt-related transcription factor 2 (RUNX-2) expressing osteoblastic cells of the endosteal niche [10]. Parathyroid hormone treatment increases the number of metastases due to its ability to promote osteoblast differentiation [10]. The presence of annexin A2 in the endosteal niche was shown to induce dormancy by upregulating growth arrest-specific 6 (GAS6) ligand of the TAM family of receptor tyrosine kinases, like AXL and TYRO3 [19]. GAS6 production was derived from osteoblasts and prevented prostate cancer cell proliferation in vitro. This was shown to be mediated by up-regulation transforming growth factor-β (TGF-β) and TGF-β receptor (TGF-βR) [20]. Further studies with acute lymphoblastic leukemia (ALL) cells showed that extracellular osteopontin within the endosteal niche led to induction of dormancy and cell cycle arrest [21].
Role of hypoxia
Tumor progression is strongly influenced by the interactions of the tumor cells with its TME [1]. Based on the TME and the interplay with the niche, the primary tumor may metastasize, be eradicated, or establish dormant micro-metastases. Most solid tumors present with a disruption in oxygen homeostasis leading to a hypoxic TME [55]. This hypoxic environment is a major driving force for tumor dormancy and gives rise to a subpopulation of DTCs that are programmed to become dormant [56]. A study that exposed MDA-MB-231 breast cancer cells to intermittent, chronic hypoxia in vitro entered a dormant state, as noted by their entry into G0-G1 cell cycle arrest [57]. In vivo, these cells were also exposed to hypoxic conditions and began to demonstrate markers of dormancy and an increased prevalence of entry into tumor cell dormancy [58]. Additionally, it is postulated that hypoxia can contribute to cancer dormancy through different pathways that may be interconnected, i.e., epithelial-mesenchymal transition (EMT), especially as these cells adapt to the environment, which is regulated by over a hundred genes. An up-regulation of TGF-β2 and proliferation inhibitor p27 expression in bone marrow induced by hypoxia has been shown in dormant cancer cells, especially in head and neck cancer studies [59]. In contrast, low levels of TGF-β2 in the lung also enabled metastatic outgrowth. Hypoxia-inducible factor (HIF) is a transcription factor that is known to play a major role in the acute hypoxic response of cancer cells [60]. In these hypoxic TMEs, the use of energy resources like oxygen, ATP, and glucose is decreased in the dormant state as cellular activity suppression is beneficial for cancer cell survival. Hypoxia-inducible gene domain family member 1A (HIGD1A) is usually expressed near the necrotic region of solid tumors [61]. Under glucose-deprived conditions (i.e., chronic hypoxia), the HIGD1A promoter is demethylating, inducing HIGD1A. This activates AMPK, suppresses oxidative phosphorylation, and decreases the production of reactive oxygen species (ROS). Therefore, HIGD1A supports cancer cell survival under hypoxic conditions. Endo et al studied a pancreatic cancer cell line, AsPC-1, kept dormant in a chronic hypoxic TME [62]. Oxygen and glucose consumption, ATP turnover, and lactate production were all decreased. Dormancy was induced by AKT suppression, while AsPC-1 that were not AKT suppressed exhibited non-dormant stages and had continual consumption of energy sources, and died under the chronic hypoxic TME. The PI3K/AKT pathway has been shown to be suppressed in the induction of tumor dormancy in vivo [22]. This pathway is frequently activated in many cancers, as it regulates cell proliferation, survival, and metabolism. However, AKT suppression is needed to preserve the energy source and decrease the metabolic demand for cancer cells so that they can survive in any TME. In dormant head and neck squamous cell carcinoma (HNSCC), it was found that AKT phosphorylation was reduced, further showing that AKT signaling was suppressed [63]. These cells did still maintain mTOR activation through the up-regulation of ras homolog enriched in the brain (RHEB) and activating transcription factor 6α (ATF6α), which protected these dormant cells from apoptosis. This showed that dormant DTCs can rewire specific pathways to coordinate homeostasis and survival during growth arrest, showing an added step that must be considered in therapy. One way that AKT signaling was seen to be suppressed was in tumor cells that detached from the extracellular matrix and induced autophagy along with an antioxidant response [64]. In ovarian cancer cell lines, floating tumor spheroid cells that were isolated from the patient’s ascites fluid were seen to be in mitotic arrest due to AKT inhibition and increased p130 and p27 protein levels. When the same cells were reattached to their extracellular matrix in the ovaries, AKT was re-activated and the quiescent cells entered a proliferative state again. This helped understand whether dormancy is the property of cells suspended in the peritoneum or just of cells in specific anatomical sites, especially in cancers like ovarian cancer, where metastasis occurs in the omentum and mesothelium [65].
Role of autophagy
Another proposed mechanism of survival during dormancy involves autophagy, which allows for dormant cells to maintain metabolic fitness, despite being in a growth-arrested environment. Lu et al showed that autophagy is induced in dormant ovarian tumors in mice, as shown by the up-regulation in the expression of autophagy-related genes (ATG) [66]. The use of chloroquine to inhibit autophagy in these dormant tumor cells resulted in a decrease in tumor regrowth, showing that autophagy is likely a survival mechanism for dormant cells and helps tumor cells survive certain targeted therapies. A study with mouse transgenic gastrointestinal stromal tumor cells showed a survival to imatinib (KIT and BCR-ABL inhibitor) treatment only when a reversible quiescent phenotype was displayed with elevated ATG7 and ATG12 levels [67]. The autophagy signaling machinery that is regulated by the ATG genes is shown to help integrate quiescence with survival signals, promoting damage repair and generating an alternate route for ATP generation through amino turnover.
Role of immunosurveillance
The immune cells are a key part of the TME. A case report in 1998 showed that a prior melanoma patient donated their kidney, which later developed melanoma metastases within the transplanted kidney of the donor-recipient [68]. This suggested that remaining melanoma cells were dormant within the donor’s body due to the immune system and escaped dormancy and progressed to metastasis upon entering a new immune system. The mechanism behind this is unclear and could either be attributed to a new immune system or due to a new TME. In a sarcoma model, stable tumor masses remained in a static state for more than 5 months before spontaneous regression. During this static state, the apoptotic fraction was high and the Ki67 index, a determinant of G0 exit, was low, indicating the involvement of CD8+ T-cell-mediated cytotoxicity. Upon CD8+ and CD4+ T-cell depletion, the tumor escaped its static state and resumed proliferation, indicating the involvement of adaptive immunity in tumor mass dormancy [69]. In a study involving the pancreatic RIP-Tag2 cancer mouse model, injection of CD4+ T cells arrested tumor progression, independently of CD8+ T cells through interferon-γ (IFN-γ) and tumor necrosis factor receptor (TNFR1) signaling. These CD4+ T-cell effects were seen due to the involvement of CXCL9 and CXCL10, angiogenesis inhibitors. This indicated the involvement of T cells with endothelial cells and angiogenesis inhibition [70]. Another study showed that IFN-γ derived from T helper cells helps to induce an irreversible state of tumor cell senescence in T antigen-induced pancreatic cancer models [71]. Overall, immune-mediated changes in TME show involvement in the induction and escape of tumor dormancy, but a mechanistic understanding is yet to be completely understood. Some individual metastatic cancer cells have shown to not express major histocompatibility complex I (MHC I) and tumor antigens [72]. Pancreatic ductal carcinomas recently displayed a new immune escape mechanism that was used by dormant cells. Endoplasmic reticulum stress for DTCs in the liver did not express tumor antigen cytokeratin 19 and MHC I, which allowed for evasion of T-cell recognition and killing [73]. Several studies with multiple myeloma cells have suggested a theory of “immunocloaking”, where the multiple myeloma cell can disguise itself as a cell that belongs to the niche, rather than as an invader [48]. Hereby, dormant multiple myeloma cells can be disguised as osteomacs or CD169+ bone marrow macrophages, which are known to reside within the endosteal niche [74]. This theory is distinct from the convention immune evasion methods that were originally studied in cancer. In immunocloaking, MHC II can engage lymphocyte activation gene 3 (LAG3) and Fc receptor-like protein 6 (FCRL6) on cytotoxic natural killer cells, CD4+ and CD8+ T cells, which synergistically suppresses immunity against MHC II expressing cancer cells [75-77]. Studies showed that LAG3 engagement of MHC II protected melanoma cells from programmed cell death while also suppressing cytotoxic T-cell killing of the tumor, allowing it to survive [78].
Role of extracellular matrix
The extracellular matrix is an important part of the TME where the ratio of ERK and p38MAPK attachment to the extracellular matrix is the molecular switch for cellular dormancy [79]. A study was performed with human epidermoid carcinoma HEp3 cells, where proliferation was shown to be regulated by suppression of p38MAPK by fibronectin and the activation of ERK by urokinase-plasminogen activator receptor (uPAR) and α5β1 integrin [80]. Any imbalance between this suppression and activation leads to dormancy in HEp3 cells due to growth suppression from ERK and p38MAPK. Blockage of p38MAPK showed to alter the imbalance and extricate the cells from dormancy and resume proliferation. Endoplasmic reticulum stress, due to impaired protein folding in hypoxic conditions, plays a role in dormancy induction as well [81]. White et al [82] reported that inhibition of β1 integrin was able to induce dormancy in vitro and in vivo in mouse breast cancer cell lines. The cell lines show that activation of β1 integrin was able to produce fibronectin and actin stress fibers, showing the importance of the extracellular matrix and the TME in cellular dormancy. DTC integrin binding to the perivascular niche, especially its extracellular matrix and cell surface receptors, may also induce chemoresistance along with dormancy [23].
Role of angiogenesis
Angiogenesis is a required function for tumor survival. If the tumor tissues fail to gain access to the vasculature, apoptosis occurs and a balance between proliferation and apoptosis can occur to put tumor cells into a clinically dormant state [83]. A recent study with breast cancer DTCs showed that these cells were inhabiting on microvasculature in their respective metastatic organs within the perivascular niche [9]. The microvasculature was studied and was producing thrombospondin, which is associated with keeping tumor cells in a dormant state. However, the tip cells of the neovasculature were shown to produce TGFβ1 and periostin, allowing for these solitary cells to initiate proliferation as needed. This shows that thrombospondin works as an angiogenesis regulator that can control a vascular switch between dormancy and proliferation. The “angiogenic switch” is referred to the transition of a pre-vascular lesion to a highly vascularized tumor that is able to progressively grow [84]. Therefore, tumors unable to successfully gain blood vessel access remain in a dormant, quiescent state until they acquire enough mutations to induce the angiogenic switch and escape from dormancy into a proliferative state. The addition of vascular endothelial growth factor (VEGF) to human melanoma cells showed enough growth to escape tumor dormancy [85]. A study performed on mice showed that epoxyeicosatrienoic acids in the endothelium of blood vessels worked as an angiogenesis inducer and stimulated exit from a dormant state [69]. Epoxyeicosatrienoic acids were then shown to help stimulate metastasis of various xenograft tumors, including Lewis lung carcinomas and B16-F10 melanomas [86]. In situ imaging of melanoma and breast cancer DTCs in mouse brains showed that dormant cells survived by establishing perivascular localization and endothelial contact [87]. The studies on angiogenic dormancy seem to point towards the belief that decreased oxygenation in the microenvironment is a more dominant driver of cells into dormancy.
Factors involved in the escape from dormancy
While mechanisms for tumor dormancy induction and quiescence maintenance have been studied, the mechanisms for escape from dormancy into recurrence of the primary tumor have also been investigated. CD36 is a fatty acid translocase macrophage scavenger receptor that can bind thombospondin 1 and collagen, which is expressed on metastatic human oral cancer cells and prostatic cancer cells [88, 89]. CD36 takes up fatty acids and fuels lipid β-oxidation, which allows for growth of metastatic tumor [90]. Further studies of CD36’s effects showed that dietary fat intake had a large effect on the ability of β-oxidation to occur while neutralizing antibodies to CD36 also prevented the further development of tumor metastases [89]. It is still not clear whether dormant cancer cells truly require CD36 for niche survival. However, other lipid mediators have been implicated in some studies to be involved in cancer dormancy escape. Cytoskeletal architecture and the extracellular matrix properties are shown to be involved in dormancy escape. In metastatic breast cancer, dormant cells escaped into a proliferative state via periostin activation, which is produced by endothelial tip cells and fibroblasts [9]. Another study showed metastatic breast cancer cell lines lying dormant in bone marrow escaped from dormancy upon upregulation of vascular cell adhesion protein 1 (VCAM 1) [24]. A reduction of breast cancer metastasis to the lung was seen by inducing dormancy of the DTC in the lung upon myosin light chain kinase (MLCK) inhibition, showing that MLCK is involved in the switch from dormancy to proliferation. Fibronectin and β1 integrin were shown to activate MLCK, maintaining a proliferative state over a dormant state [47]. The involvement of the angiogenic switch and epoxyeicosanoids generated from arachidonic acid has also been implicated in tumor escape in some studies [86]. In the lung, inflammation led to neutrophil extracellular traps (NETs), which formed scaffolds of chromatin and proteolytic enzymes, which remodeled the extracellular matrix and its control of tumor cell dormancy, leading to reactivation of dormant breast cancer cells with lung metastases [91]. This has demonstrated the displacement of dormant cells from their niche. A multiple myeloma model demonstrated this where receptor activator of nuclear factor-κB (NF-κB) ligand (RANKL) stimulated osteoclast resorption and decreased the number of dormant myeloma cells [8]. Another study with dormant mouse breast and prostate cancer cells was identified and showed induction of bone resorption by ovariectomy/castration, which leads to increased bone metastasis formation [92]. Bone metastasis, in this case, was blocked by bisphosphonates and RANKL inhibitors, both being osteoclast inhibitors [93, 94]. Stimulation of osteoclastic bone resorption cascades into a cycle where the cancer cells secrete a parathyroid hormone-related protein (PTHRP), which leads to more osteoclastic bone resorption, which releases mediators like TGF-β1 which further induces more PTHRP secretion, which further continues the cycle, leading to amplification of metastasis [95].
Advances in targeting DTCs
Kondo et al have made advances in forming a platform for studying cellular dormancy of cancer in vitro by developing the cancer tissue-originated spheroid (CTOS) method. In this method, dormancy can be induced in hypoxia for at least 7 days without growth factor stimulation. Studies with CTOS have shown that cellular dormancy is a feature commonly known to cancer cells, and not restricted specifically to certain cell lines. Additionally, researchers have been using lentiviral vectors to introduce reporter proteins, like fluorescent fusion proteins and luciferase reporters, to help mark non-proliferating cells and tag cells in G0 cell cycle arrest [23]. Recent studies and utilization of new techniques have all shown one major theme: dormant cancer cells localize to their niches in specific microanatomical locations. For example, multiple myeloma [8], ALL cells [21], and prostate cancer cells [10] have been shown to localize to the endosteal surface of bone while other cancers have been shown to localize perivascular regions of various organs [9, 17]. Further studies that model these dormant cancer cells in their niches in vivo will be crucial in elucidating more information on cancer dormancy. New technologies such as in situ photolabeling and spatial transcriptomics show promise in learning more about these niches, especially in bone and other metastatic organs [25, 26]. Recent progress in intravital imaging and RNA-sequencing has allowed for studies to target specific stages in the dormant cancer cell life cycle with specific cell cycle therapies to eradicate and prevent metastases [27]. The various mechanisms involved in tumor dormancy are illustrated in Figure 1.
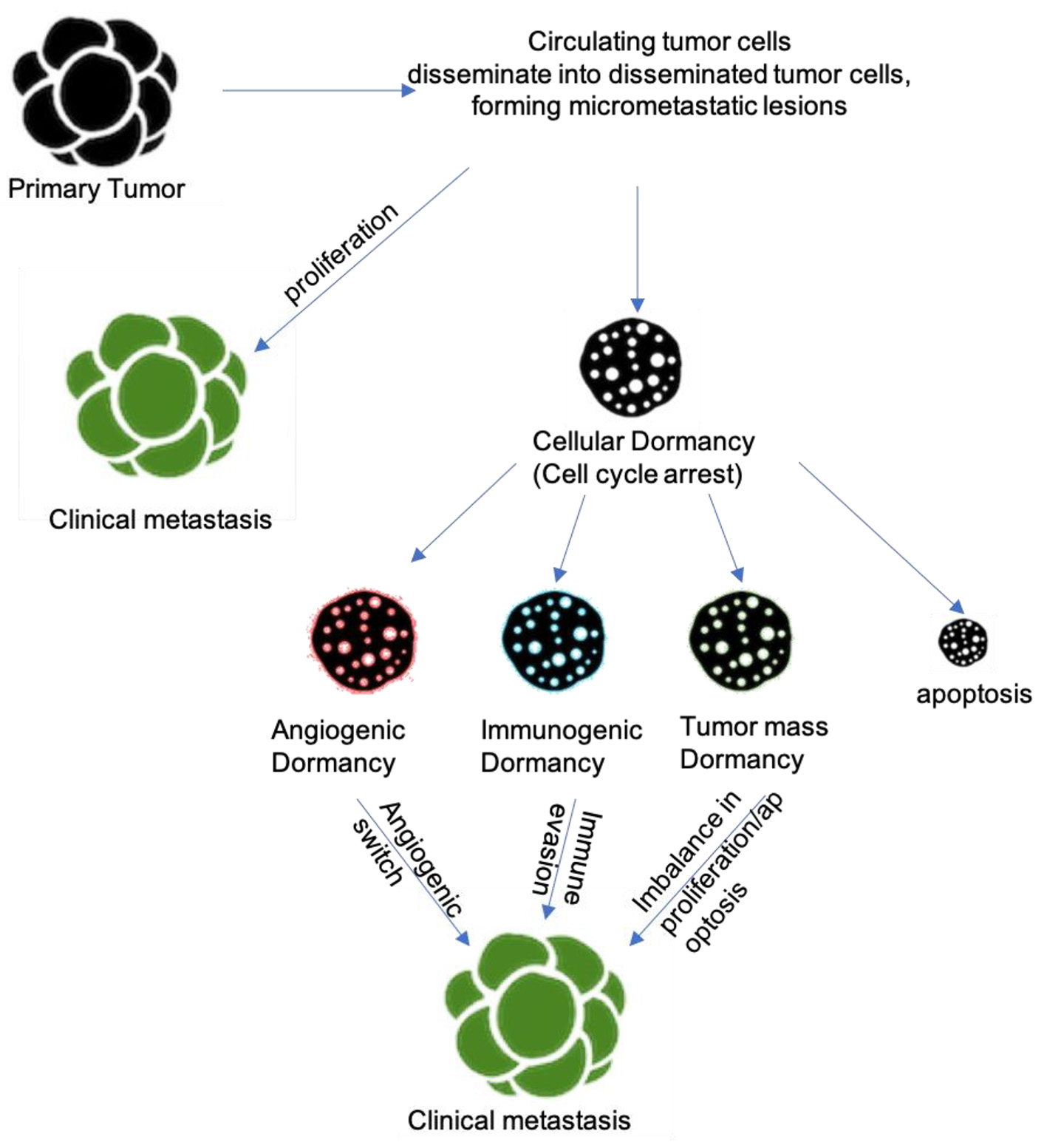 Click for large image | Figure 1. Mechanisms/pathways underlying tumor dormancy. |
Advances have been made by studying drug-induced dormancy and seeing how blocking survival signals during this phase affect tumor growth and dissemination. In multiple myeloma cells, bortezomib has been shown to induce quiescence [28]. Salubrinal (GADD34-PP1c inhibitor) treatment to these bortezomib-treated tumor cells resulted in induction of apoptosis in the quiescent cells, showing potential for future therapy by inducing quiescence and then inducing apoptosis in those quiescent cells. For these specific multiple myeloma cells, eukaryotic translation initiation factor 2α (EIF2α) dephosphorylation inhibition was achieved by salubrinal, which allowed the EIF2α to remain hyperphosphorylated, allowing bortezomib-induced apoptosis to occur. This shows its potential as a novel therapeutic approach that could reduce residual disease and recurrences in multiple myeloma [28]. A similar approach was used in a study with non-small-cell lung cancer (NSCLC) xenografts which were treated with erlotinib, an EGFR inhibitor [29]. These cells entered a quiescent state upon erlotinib treatment but were killed when this therapy was combined with ABT-737, a BCL-2, and a BCL-XL inhibitor. BCL-2 and BCL-XL are stress-related proteins, whose regulation promotes survival in a quiescent state.
Targeting the process of dormant cancer cell reprogramming and niche adaptation shows promise for therapy in the future. A study using 5-azacytidine with retinoic acid receptor agonists reinstated NR2F1-induced dormancy [30]. This may be suggestive of the possibility of inducing dormancy in metastatic cells. Current studies are now testing this idea in prostate cancer cells. Additionally, studies implementing hypoxia inhibition and glycolysis inhibition and their impact on dormant cells within their niches show promise and could also be later implemented as a preventative relapse measure [31, 32]. Originally, targeting genes that are differentially expressed by dormant cancer cells showed potential for therapy. However, many cases showed that dormant cell surface molecules like AXL and VCAM1 were also expressed by macrophages, making it likely that targeting these molecules would interfere with immunity and be unsuccessful [33]. However, with the introduction of the concept of “immunocloaking” in multiple myeloma cells, the idea is that “un-cloaking” these disguised cells may allow for their elimination. Several clinical trials testing relatlimab, a LAG3 monoclonal antibody, show promise in being able to further confirm or deny this hypothesis [34]. The LAG3 ligand is expressed on MHC II and data suggest that dormant cancer cells may express MHC II in high levels, allowing resistance to immune checkpoint inhibition [35].
Another mechanism of recent studies has attempted to target the cell cycle of dormant cancer cells and keep it an arrest. However, the use of mechanisms like CDK inhibitors has not yielded much success [36]. However, preclinical data are showing that micro-environment-dependent mechanisms that control reactivation of dormancy could be promising: targeting osteoclastic bone resorption, which releases cells from dormancy, shows promise [24]. Bisphosphonate zoledronic acid is known to inhibit bone resorption [37]. The data from the AZURE trial are showing that administration of bisphosphonate zoledronic acid decreases metastases development in breast cancer patients [38]. The data are further supported by the fact that bisphosphonate treatment has been shown to improve survival in postmenopausal breast cancer patients [96]. It is well known that the detection of dormant lesions can have significant implications and both the early treatment and prevention of cancer. For breast cancer specifically, it is estimated that nearly a third of women aged 40 - 50 years may have clinically undetectable breast cancer [39]. Detection of dormant micrometastatic lesions could lead to better detection and early treatment for a better prognosis. Additionally, tumor late recurrences and long, undetectable periods of minimal residual disease are also associated with tumor dormancy and its mechanisms. Currently, we are still challenged by the inability to detect clinically occult primary tumors or micro-metastases [5]. Blocking tumors in their early quiescent stages could likely result in better patient outcomes over tumors that are highly vascularized and large with systemic symptoms. This dormant stage of tumors may be a very promising therapeutic target in the future. Future studies must begin considering approaches within clinical trials of new agents to understand the implications of these agents on dormant cancer cells and DTCs within the niche in which they reside.
Prior therapeutic trials have emphasized taking tumor cells out of their quiescent state and then killing them, but showed to have a worse patient outcome [40]. However, if these quiescent cells could be eradicated while dormant, this would be a new strategy to prevent metastasis in these dormant situations [41]. However, more information on the mechanisms behind dormancy must be gained before this strategy can be implemented clinically. Residual DTCs are usually genetically heterogeneous and if these cells were awakened from dormancy, the genetic repertoire would expand, allowing for therapy resistance [22]. Molecular characterization of ncRNAs as potential biomarkers in human cancers may contribute to our understanding of tumor dormancy and lead to more future therapies. More biomarkers of dormancy can be developed and may help detect and treat tumors and recurrent DTCs before they escape dormancy and become proliferative.
| Conclusions | ▴Top |
The detection and understanding of the concept of tumor dormancy have significantly improved in the last two decades. Extensive molecular and genetic characterization of disseminated and circulating tumor cells has contributed to our understanding of the frequency and prevalence of tumor dormancy. While the exact mechanisms are unknown, we have seen that dormancy is induced and maintained through an interplay of TME changes, including hypoxia, angiogenic mechanisms, and immune mechanisms. More work is to be done in further understanding the mechanisms that lie behind this concept. Upon more elucidation of this, therapeutic treatments have the potential to be very beneficial for patients. At this time, there is no treatment available that can keep tumors in a dormant, cell-cycle arrested, and asymptomatic state for a prolonged period. However, the understanding that several humans carry microscopic cancerous lesions without experiencing any symptoms implies that our bodies have some inherent defense mechanisms against full-blown tumor development that are overcome rarely. Gaining a further understanding of the process by which a tumor overcomes these dormant growth restrictions and emerges into a proliferative state can lead to the creation of some novel strategies that prolong dormancy or block the switch from dormancy to proliferation, or even metastasis if detected at an early enough stage.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Author Contributions
Vanshika Balayan performed research, analyzed data, and wrote the paper. Achuta Guddati designed research, performed research, analyzed data, and wrote the paper.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Butturini E, Carcereri de Prati A, Boriero D, Mariotto S. Tumor dormancy and interplay with hypoxic tumor microenvironment. Int J Mol Sci. 2019;20(17):4305.
doi pubmed - Friberg S, Nystrom A. Cancer metastases: early dissemination and late recurrences. Cancer Growth Metastasis. 2015;8:43-49.
doi pubmed - Hadfield G. The dormant cancer cell. Br Med J. 1954;2(4888):607-610.
doi pubmed - Cox TR, Bird D, Baker AM, Barker HE, Ho MW, Lang G, Erler JT. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013;73(6):1721-1732.
doi pubmed - Almog N. Molecular mechanisms underlying tumor dormancy. Cancer Lett. 2010;294(2):139-146.
doi pubmed - Wikman H, Vessella R, Pantel K. Cancer micrometastasis and tumour dormancy. APMIS. 2008;116(7-8):754-770.
doi pubmed - Townson JL, Chambers AF. Dormancy of solitary metastatic cells. Cell Cycle. 2006;5(16):1744-1750.
doi pubmed - Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, Kaplan W, et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun. 2015;6:8983.
doi pubmed - Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, Almeida D, et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol. 2013;15(7):807-817.
doi pubmed - Shiozawa Y, Pedersen EA, Havens AM, Jung Y, Mishra A, Joseph J, Kim JK, et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest. 2011;121(4):1298-1312.
doi pubmed - Vora AJ, Toh CH, Peel J, Greaves M. Use of granulocyte colony-stimulating factor (G-CSF) for mobilizing peripheral blood stem cells: risk of mobilizing clonal myeloma cells in patients with bone marrow infiltration. Br J Haematol. 1994;86(1):180-182.
doi pubmed - Nahi H, Celanovic M, Liu Q, Lund J, Peceliunas V. A pilot, exploratory, randomized, phase II safety study evaluating tumor cell mobilization and apheresis product contamination in patients treated with granulocyte colony-stimulating factor alone or plus plerixafor. Biol Blood Marrow Transplant. 2019;25(1):34-40.
doi pubmed - Nie Y, Han YC, Zou YR. CXCR4 is required for the quiescence of primitive hematopoietic cells. J Exp Med. 2008;205(4):777-783.
doi pubmed - McCabe NP, De S, Vasanji A, Brainard J, Byzova TV. Prostate cancer specific integrin alphavbeta3 modulates bone metastatic growth and tissue remodeling. Oncogene. 2007;26(42):6238-6243.
doi pubmed - Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62(6):1832-1837.
- Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410(6824):50-56.
doi pubmed - Price TT, Burness ML, Sivan A, Warner MJ, Cheng R, Lee CH, Olivere L, et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci Transl Med. 2016;8(340):340ra373.
doi - Shiozawa Y, Havens AM, Jung Y, Ziegler AM, Pedersen EA, Wang J, Wang J, et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J Cell Biochem. 2008;105(2):370-380.
doi pubmed - Shiozawa Y, Pedersen EA, Patel LR, Ziegler AM, Havens AM, Jung Y, Wang J, et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia. 2010;12(2):116-127.
doi pubmed - Yumoto K, Eber MR, Wang J, Cackowski FC, Decker AM, Lee E, Nobre AR, et al. Axl is required for TGF-beta2-induced dormancy of prostate cancer cells in the bone marrow. Sci Rep. 2016;6:36520.
doi pubmed - Boyerinas B, Zafrir M, Yesilkanal AE, Price TT, Hyjek EM, Sipkins DA. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood. 2013;121(24):4821-4831.
doi pubmed - Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14(9):611-622.
doi pubmed - Carlson P, Dasgupta A, Grzelak CA, Kim J, Barrett A, Coleman IM, Shor RE, et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat Cell Biol. 2019;21(2):238-250.
doi pubmed - Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, Yan J, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell. 2011;20(6):701-714.
doi pubmed - Nguyen A, Khoo WH, Moran I, Croucher PI, Phan TG. Single Cell RNA Sequencing of Rare Immune Cell Populations. Front Immunol. 2018;9:1553.
doi pubmed - Burgess DJ. Spatial transcriptomics coming of age. Nat Rev Genet. 2019;20(6):317.
doi pubmed - Ghajar CM. Metastasis prevention by targeting the dormant niche. Nat Rev Cancer. 2015;15(4):238-247.
doi pubmed - Schewe DM, Aguirre-Ghiso JA. Inhibition of eIF2alpha dephosphorylation maximizes bortezomib efficiency and eliminates quiescent multiple myeloma cells surviving proteasome inhibitor therapy. Cancer Res. 2009;69(4):1545-1552.
doi pubmed - Fan W, Tang Z, Yin L, Morrison B, Hafez-Khayyata S, Fu P, Huang H, et al. MET-independent lung cancer cells evading EGFR kinase inhibitors are therapeutically susceptible to BH3 mimetic agents. Cancer Res. 2011;71(13):4494-4505.
doi pubmed - Sosa MS. Dormancy programs as emerging antimetastasis therapeutic alternatives. Mol Cell Oncol. 2016;3(1):e1029062.
doi pubmed - Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25(34):4633-4646.
doi pubmed - Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11(6):393-410.
doi pubmed - Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, Ekpin E, et al. NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nat Commun. 2015;6:6170.
doi pubmed - Johnson DB, Nixon MJ, Wang Y, Wang DY, Castellanos E, Estrada MV, Ericsson-Gonzalez PI, et al. Tumor-specific MHC-II expression drives a unique pattern of resistance to immunotherapy via LAG-3/FCRL6 engagement. JCI Insight. 2018;3(24):e120360.
doi pubmed - Khoo WH, Ledergor G, Weiner A, Roden DL, Terry RL, McDonald MM, Chai RC, et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood. 2019;134(1):30-43.
doi pubmed - Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14(2):130-146.
doi pubmed - Coleman RE, Collinson M, Gregory W, Marshall H, Bell R, Dodwell D, Keane M, et al. Benefits and risks of adjuvant treatment with zoledronic acid in stage II/III breast cancer. 10 years follow-up of the AZURE randomized clinical trial (BIG 01/04). J Bone Oncol. 2018;13:123-135.
doi pubmed - Coleman R, Cameron D, Dodwell D, Bell R, Wilson C, Rathbone E, Keane M, et al. Adjuvant zoledronic acid in patients with early breast cancer: final efficacy analysis of the AZURE (BIG 01/04) randomised open-label phase 3 trial. Lancet Oncol. 2014;15(9):997-1006.
doi - Black WC, Welch HG. Advances in diagnostic imaging and overestimations of disease prevalence and the benefits of therapy. N Engl J Med. 1993;328(17):1237-1243.
doi pubmed - Essers MA, Trumpp A. Targeting leukemic stem cells by breaking their dormancy. Mol Oncol. 2010;4(5):443-450.
doi pubmed - Sosa MS, Avivar-Valderas A, Bragado P, Wen HC, Aguirre-Ghiso JA. ERK1/2 and p38alpha/beta signaling in tumor cell quiescence: opportunities to control dormant residual disease. Clin Cancer Res. 2011;17(18):5850-5857.
doi pubmed - Kleinsmith LJ, Pierce GB, Jr. Multipotentiality of single embryonal carcinoma cells. Cancer Res. 1964;24:1544-1551.
- Pierce GB, Speers WC. Tumors as caricatures of the process of tissue renewal: prospects for therapy by directing differentiation. Cancer Res. 1988;48(8):1996-2004.
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755-768.
doi pubmed - Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983-3988.
doi pubmed - Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17(3):313-319.
doi pubmed - Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ, Liu ZY, et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008;68(15):6241-6250.
doi pubmed - Phan TG, Croucher PI. The dormant cancer cell life cycle. Nat Rev Cancer. 2020;20(7):398-411.
doi pubmed - Endo H, Inoue M. Dormancy in cancer. Cancer Sci. 2019;110(2):474-480.
doi pubmed - Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429-4434.
doi pubmed - Davies C, Pan H, Godwin J, Gray R, Arriagada R, Raina V, Abraham M, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2013;381(9869):805-816.
doi - Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, Peto R, et al. 20-Year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N Engl J Med. 2017;377(19):1836-1846.
doi pubmed - Sosnoski DM, Norgard RJ, Grove CD, Foster SJ, Mastro AM. Dormancy and growth of metastatic breast cancer cells in a bone-like microenvironment. Clin Exp Metastasis. 2015;32(4):335-344.
doi pubmed - Freedland SJ, Moul JW. Prostate specific antigen recurrence after definitive therapy. J Urol. 2007;177(6):1985-1991.
doi pubmed - McNichols DW, Segura JW, DeWeerd JH. Renal cell carcinoma: long-term survival and late recurrence. J Urol. 1981;126(1):17-23.
doi - Dou J, Gu N. Emerging strategies for the identification and targeting of cancer stem cells. Tumour Biol. 2010;31(4):243-253.
doi pubmed - Carcereri de Prati A, Butturini E, Rigo A, Oppici E, Rossin M, Boriero D, Mariotto S. Metastatic breast cancer cells enter into dormant state and express cancer stem cells phenotype under chronic hypoxia. J Cell Biochem. 2017;118(10):3237-3248.
doi pubmed - Fluegen G, Avivar-Valderas A, Wang Y, Padgen MR, Williams JK, Nobre AR, Calvo V, et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat Cell Biol. 2017;19(2):120-132.
doi pubmed - Bragado P, Estrada Y, Parikh F, Krause S, Capobianco C, Farina HG, Schewe DM, et al. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat Cell Biol. 2013;15(11):1351-1361.
doi pubmed - Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721-732.
doi pubmed - Ameri K, Jahangiri A, Rajah AM, Tormos KV, Nagarajan R, Pekmezci M, Nguyen V, et al. HIGD1A Regulates Oxygen Consumption, ROS Production, and AMPK Activity during glucose deprivation to modulate cell survival and tumor growth. Cell Rep. 2015;10(6):891-899.
doi pubmed - Endo H, Okuyama H, Ohue M, Inoue M. Dormancy of cancer cells with suppression of AKT activity contributes to survival in chronic hypoxia. PLoS One. 2014;9(6):e98858.
doi pubmed - Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci U S A. 2008;105(30):10519-10524.
doi pubmed - Avivar-Valderas A, Bobrovnikova-Marjon E, Alan Diehl J, Bardeesy N, Debnath J, Aguirre-Ghiso JA. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene. 2013;32(41):4932-4940.
doi pubmed - Correa RJ, Peart T, Valdes YR, DiMattia GE, Shepherd TG. Modulation of AKT activity is associated with reversible dormancy in ascites-derived epithelial ovarian cancer spheroids. Carcinogenesis. 2012;33(1):49-58.
doi pubmed - Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, Kondo S, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest. 2008;118(12):3917-3929.
doi pubmed - Gupta A, Roy S, Lazar AJ, Wang WL, McAuliffe JC, Reynoso D, McMahon J, et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc Natl Acad Sci U S A. 2010;107(32):14333-14338.
doi pubmed - MacKie RM, Reid R, Junor B. Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N Engl J Med. 2003;348(6):567-568.
doi pubmed - Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450(7171):903-907.
doi pubmed - Muller-Hermelink N, Braumuller H, Pichler B, Wieder T, Mailhammer R, Schaak K, Ghoreschi K, et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell. 2008;13(6):507-518.
doi pubmed - Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, Schilbach K, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature. 2013;494(7437):361-365.
doi pubmed - Pantel K, Schlimok G, Kutter D, Schaller G, Genz T, Wiebecke B, Backmann R, et al. Frequent down-regulation of major histocompatibility class I antigen expression on individual micrometastatic carcinoma cells. Cancer Res. 1991;51(17):4712-4715.
- Pommier A, Anaparthy N, Memos N, Kelley ZL, Gouronnec A, Yan R, Auffray C, et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science. 2018;360(6394):eaao4908.
doi pubmed - Ehninger A, Trumpp A. The bone marrow stem cell niche grows up: mesenchymal stem cells and macrophages move in. J Exp Med. 2011;208(3):421-428.
doi pubmed - Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, Auffray C, et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J Exp Med. 1992;176(2):327-337.
doi pubmed - Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, Vilgelm AE, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun. 2016;7:10582.
doi pubmed - Schreeder DM, Cannon JP, Wu J, Li R, Shakhmatov MA, Davis RS. Cutting edge: FcR-like 6 is an MHC class II receptor. J Immunol. 2010;185(1):23-27.
doi pubmed - Hemon P, Jean-Louis F, Ramgolam K, Brignone C, Viguier M, Bachelez H, Triebel F, et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol. 2011;186(9):5173-5183.
doi pubmed - Aguirre Ghiso JA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol. 1999;147(1):89-104.
doi pubmed - Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. 2001;12(4):863-879.
doi pubmed - Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006;66(3):1702-1711.
doi pubmed - White DE, Kurpios NA, Zuo D, Hassell JA, Blaess S, Mueller U, Muller WJ. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell. 2004;6(2):159-170.
doi pubmed - Holmgren L, O'Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1(2):149-153.
doi pubmed - Baeriswyl V, Christofori G. The angiogenic switch in carcinogenesis. Semin Cancer Biol. 2009;19(5):329-337.
doi pubmed - Bayko L, Rak J, Man S, Bicknell R, Ferrara N, Kerbel RS. The dormant in vivo phenotype of early stage primary human melanoma: termination by overexpression of vascular endothelial growth factor. Angiogenesis. 1998;2(3):203-217.
doi pubmed - Panigrahy D, Edin ML, Lee CR, Huang S, Bielenberg DR, Butterfield CE, Barnes CM, et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Invest. 2012;122(1):178-191.
doi pubmed - Kienast Y, von Baumgarten L, Fuhrmann M, Klinkert WE, Goldbrunner R, Herms J, Winkler F. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med. 2010;16(1):116-122.
doi pubmed - Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R, Porter LH, et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med. 2019;11(478):eaau5758.
doi pubmed - Pascual G, Avgustinova A, Mejetta S, Martin M, Castellanos A, Attolini CS, Berenguer A, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541(7635):41-45.
doi pubmed - Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, Cao Y. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer. 2017;16(1):76.
doi pubmed - Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, Upadhyay P, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361:6409).
doi pubmed - Ottewell PD, Wang N, Brown HK, Reeves KJ, Fowles CA, Croucher PI, Eaton CL, et al. Zoledronic acid has differential antitumor activity in the pre- and postmenopausal bone microenvironment in vivo. Clin Cancer Res. 2014;20(11):2922-2932.
doi pubmed - Ottewell PD, Wang N, Brown HK, Fowles CA, Croucher PI, Eaton CL, Holen I. OPG-Fc inhibits ovariectomy-induced growth of disseminated breast cancer cells in bone. Int J Cancer. 2015;137(4):968-977.
doi pubmed - Ottewell PD, Wang N, Meek J, Fowles CA, Croucher PI, Eaton CL, Holen I. Castration-induced bone loss triggers growth of disseminated prostate cancer cells in bone. Endocr Relat Cancer. 2014;21(5):769-781.
doi pubmed - Mundy GR. Mechanisms of bone metastasis. Cancer. 1997;80(8 Suppl):1546-1556.
doi - Early Breast Cancer Trialists' Collaborative Group. Adjuvant bisphosphonate treatment in early breast cancer: meta-analyses of individual patient data from randomised trials. Lancet. 2015;386(10001):1353-1361.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.