

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 13, Number 2, April 2022, pages 84-95

The Diverse Analysis Identifies Mutated KRAS Associated With Radioresistance in Non-Small Cell Lung Cancer
Dao Qi Zhua, d, Ying Liub, d, Zhi Jian Yua, Ru Hua Zhangc, Ai Wu Lib, Feng Ying Gongb, Wei Wangb, Wei Xiaoa, e, Qin Fana, e
aSchool of Traditional Chinese Medicine, Southern Medical University, Guangzhou, Guangdong 510515, China
bNanFang Hospital, Southern Medical University, Guangzhou, Guangdong 510515, China
cState Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University Cancer Center, Guangzhou, Guangdong 510515, China
dThese authors contributed equally to this work as joint first authors.
eCorresponding Author: Wei Xiao and Qin Fan, School of Traditional Chinese Medicine, Southern Medical University, Guangzhou, Guangdong 510515, Chinaand
Manuscript submitted March 21, 2022, accepted April 18, 2022, published online April 28, 2022
Short title: Mutated KRAS and Radiation Resistance
doi: https://doi.org/10.14740/wjon1465
| Abstract | ▴Top |
Background: To analyze the relationship between V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) status and radioresistance in non-small cell lung cancer (NSCLC), we identified potential genotypic differences and pathways involved.
Methods: We retrospectively analyzed epidermal growth factor receptor (EGFR) and KRAS status in patients undergoing definitive radiotherapy for NSCLC between 2004 and 2018. Cox proportional hazard models were used to evaluate local progression-free survival (LPFS). Using clonogenic survival and measurement of γH2AX foci, we analyzed the difference in radiosensitivity between NSCLC cell lines with different KRAS status. The Cancer Genome Atlas (TCGA) analysis was used to explore the potential pathways involved.
Results: The results showed that of the 286 patients identified, 68 (24%) had local tumor progression (mean ± standard deviation (SD), 27 ± 17.4 months); of these patients, KRAS mutations were found in 14 (23%), and KRAS status was associated with LPFS. After adjusting for concurrent chemotherapy, gross tumor volume, and mutation status in multivariate analysis, KRAS mutation was associated with shorter LPFS (hazard ratio: 1.961; 95% confidence interval: 1.03 - 2.17; P = 0.032). KRAS mutation showed higher radioresistance in vitro. TCGA data showed that the ERK1/2 pathway, phosphatidylinositol I3 kinase (PI3K)/mTOR, p38 MAPK pathway, cell cycle checkpoint signaling, DNA damage, repair pathways, and EGFR/PKC/AKT pathway were differentially expressed in patients with KRAS mutations or cell lines compared with their expression in the wild-type group.
Conclusions: Diverse analyses identified that KRAS mutation was associated with radioresistance in NSCLC. KRAS mutation status may be helpful as a biomarker of radioresistance and a potential target to increase radiosensitivity.
Keywords: KRAS; Non-small cell lung cancer; Radioresistance; TCGA; Biomarker
| Introduction | ▴Top |
Non-small cell lung cancer (NSCLC) accounts for 85% of all lung cancer cases [1]. Radiation therapy, alone or combined with chemotherapy, is the standard approach for the definitive treatment of locally advanced NSCLC or early-stage disease in patients who are not candidates for surgery [2, 3]. Even when concurrent chemotherapy is used with standard radiation therapy, local-regional relapse rates are unacceptably high, ranging from 20% to 50% [2-5].
A better understanding of radiation resistance and strategies to overcome it are crucial for improving treatment outcomes in NSCLC [6]. Molecular mechanisms underlying tumor radioresistance are complex and include tumor microenvironment, DNA damage and repair, and DNA checkpoint pathways [7, 8]. In NSCLC, overexpression, or mutation of the genes for epidermal growth factor receptor (EGFR) and V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) has been linked with lung cancer prognosis [9, 10]. However, their role in radiosensitivity remains unclear.
To date, most clinical studies involving molecular biomarkers have focused on the ability of such markers to predict prognosis or be used as the basis for targeted inhibitors rather than as predictors of radiosensitivity. Few clinical studies on radiotherapy and KRAS have reported conflicting results [11-14]. Despite the limited results from clinical studies, numerous laboratory investigations have indicated that KRAS genotypes have specific properties that are expected to affect radioresistance [15-18]. Thus, we hypothesized that KRAS mutation status could predict radioresistance of a particular tumor. To test this hypothesis, we retrospectively analyzed patients with NSCLC who had received definitive radiation therapy and whose KRAS mutation status was known. We investigated potential relationships between local tumor progression and mutation status to identify KRAS as a molecular marker of radioresistance using an integrative strategy, combining the results of in vitro experiments and the Cancer Genome Atlas (TCGA) data to analyze the role of KRAS in radioresistance and the potential gene pathway.
| Materials and Methods | ▴Top |
Patient selection, local tumor progression, and follow-up
Ethics committee approval was obtained from the Medical Ethics Committee of the NanFang Hospital of Southern Medical University (number: NFEC-2017-031). The study was conducted in compliance with the ethical standards of the responsible institution on human subjects as well as with the Helsinki Declaration. Patients were selected from a clinical database of patients with NSCLC who had received definitive radiation therapy at a single institution. Inclusion criteria were as follows: 1) histologically confirmed stage I - III NSCLC; 2) receipt of ≥ 60 Gy as definitive radiotherapy (or 60 Gy (RBE)) for proton therapy); and 3) available histologic reports on tumor EGFR and KRAS status. Patients treated with stereotactic ablative radiation therapy or those with unconfirmed NCSLC, stage IV NSCLC, or small cell lung cancer were excluded. A total of 286 patients who met these criteria were identified; these patients had received radiation therapy between May 15, 2004, and April 2, 2014.
Local tumor progression was defined as disease that persisted or recurred within either the radiation field or at the margin of the field [19]. Briefly, in-field progression occurred inside the planning target volume (PTV) or within the 95% prescribed isodose volume; marginal progression occurred outside the PTV, but ≤ 1 cm from the PTV boundary, or outside the 95% specified isodose volume, but within 1 cm of the 95% isodose line. At least two experienced radiation oncologists, who reviewed radiology reports and computed tomography (CT) scans, positron emission tomography (PET) scans, or PET/CT images, confirmed progression. Biopsy was not required to confirm local progression if serial imaging revealed persistent or recurrent disease [20].
Follow-up visits were conducted at least once before radiation therapy and weekly during treatment; each visit included interval history and physical examinations. Post-treatment follow-up visits were scheduled during the first 1 - 3 months after completing radiation therapy, every 3 - 4 months thereafter for the first 2 - 3 years, and then twice a year until 5 years after completing radiation therapy. Chest CT and PET were performed every 3 - 6 months after radiation therapy.
Cell lines and reagents
Lung cancer cell lines with KRAS mutation (H460 and A549) and wild-type KRAS (H1299 and H661) were used. The cancer cell lines were cultured in RPMI-1640 medium. All experiments were performed using confluent cultures maintained in 10% serum.
Clonogenic assays
Cell lines with KRAS mutation (H460 and A549) and wild-type KRAS (H1299 and H661) were grown to 40-60% confluency; 50 cells were plated for the control (no radiation) condition with an increased number of cells plated for samples exposed to higher doses of radiation (150 for 2 Gy, 300 for 4 Gy, and 600 for 6 Gy). After irradiation, the plates were placed back into a 37 °C incubator with 5% CO2 and allowed to divide for 10 - 14 days until sufficient colonies with more than 50 cells per colony were obtained. The medium was then removed, and the cells were stained with 0.5% crystal violet (Sigma-Aldrich, St. Louis, MO, USA) in methanol, rinsed, and colonies containing more than 50 cells were counted. Survival was calculated relative to that of non-irradiated cells (survival = (plating efficiency of treated cells)/(plating efficiency of control cells), where plating efficiency = (number of colonies formed by treated cells)/(number of colonies formed by untreated cells)).
Immunofluorescence
H460 cell lines with KRAS mutation and H1299 cell lines with wild-type KRAS were grown on glass coverslips, and irradiated with 4 Gy after 1 h, 8 h, and 24 h; washed with phosphate-buffered saline (PBS); fixed in 2% paraformaldehyde/PBS for 10 min; and processed for immunofluorescence using the relevant γH2AX antibody (1:400, Cell Signaling Technology)). The relevant secondary antibodies were fluorochrome-conjugated Cy3 (1:300, Jackson ImmunoResearch). Images were captured using a digital camera (AxioCam MRm; Carl Zeiss MicroImaging, Inc.) attached to a fluorescent microscope (Axioskop2 Mot Plus; Carl Zeiss MicroImaging, Inc.) (× 100 magnification). AxioVision LE 4.3 software (Carl Zeiss MicroImaging, Inc.) was used to capture the individual images. Fluorescence intensity was quantitated using ImageJ software.
TCGA database analysis
Archived data were from The Cancer Genome Atlas for Lung Adenocarcinoma (TCGA LUAD) database (https://tcga-data.nci.nih.gov). Data were selected based on patient and cell samples subjected to reverse-phase protein array (RPPA) analysis. One hundred sixty patients and 160 cell lines were available in the database. RPPA analysis selected at the false discovery rate (FDR) level of 0.10 was used to create the heatmap for patients. For the cell lines, the top 15 were used to create the heat map. The P value (≤ 0.05) hits from the records were then collectively input into the protein association networks (http://genecodis.cnb.csic.es) to determine the pathway.
Statistical analysis
The relationship between KRAS status and other clinicopathological characteristics was analyzed using the Chi-squared test. Means of age, radiation dose, and gross tumor volume (GTV) were compared using Mann-Whitney U tests. Local progression-free survival (LPFS) was calculated from the date of definitive radiation therapy termination using the Kaplan-Meier actuarial method. The influence of variables on survival was studied using univariate and multivariate analyses (Cox proportional hazards models). Independent sample t-tests were used to compare the average number of γH2AX foci in H1299 and H460 cells after radiation at different time points. All analyses were performed using Stata version 10.1. The hazard ratios (HR) and 95% confidence intervals (CIs) were calculated. Differences were considered statistically significant at P < 0.05.
| Results | ▴Top |
Mutated KRAS increased local tumor progression after definitive radiation therapy for patients with NSCLC
The characteristics of the 286 identified patients are shown in Table 1; 68 patients (24%) had local progression, the mean (± standard deviation (SD) patient age was 63.9 (± 10.4) years, and most patients (252, 88%) had stage III disease. The progression/no progression groups were relatively well balanced, except for age (patients without progression were slightly older than those with progression, P = 0.034), radiation modality (29% of those treated with photons had progression vs. 16% of those treated with protons, P = 0.02), and receipt of induction chemotherapy (32% of those who had received induction chemotherapy vs. 19% of those who had not received induction chemotherapy, P = 0.012). Mean radiation dose was similar between patients who did and did not experience local progression (68.1 ± 5.1 Gy or Gy (RBE) vs. 68.7 ± 6.5 Gy or Gy (RBE), P = 0.785). Although the SD values were large, the GTV was not different for those who did not experience progression. Most patients (262, 94%) received concurrent chemotherapy, but the progression rate was higher among those who received induction therapy (32%) than among those who did not undergo induction chemotherapy (19%, P = 0.02). The median follow-up time for the 68 patients with local progression was 57.4 months (range 1.27 - 93.5 months), and the mean interval to progression was 27 months (± 17.4 months (SD)). Among the patients who experienced local progression, EGFR mutations were detected in five of 67 patients (7%) and KRAS mutations in 14 of 60 patients (23%) (Table 1). Only one patient had mutations in both KRAS and EGFR and was excluded from the LPFS analysis. The PET and CT images of a representative patient with local progression are shown in Figure 1.
 Click to view | Table 1. Patient Characteristics |
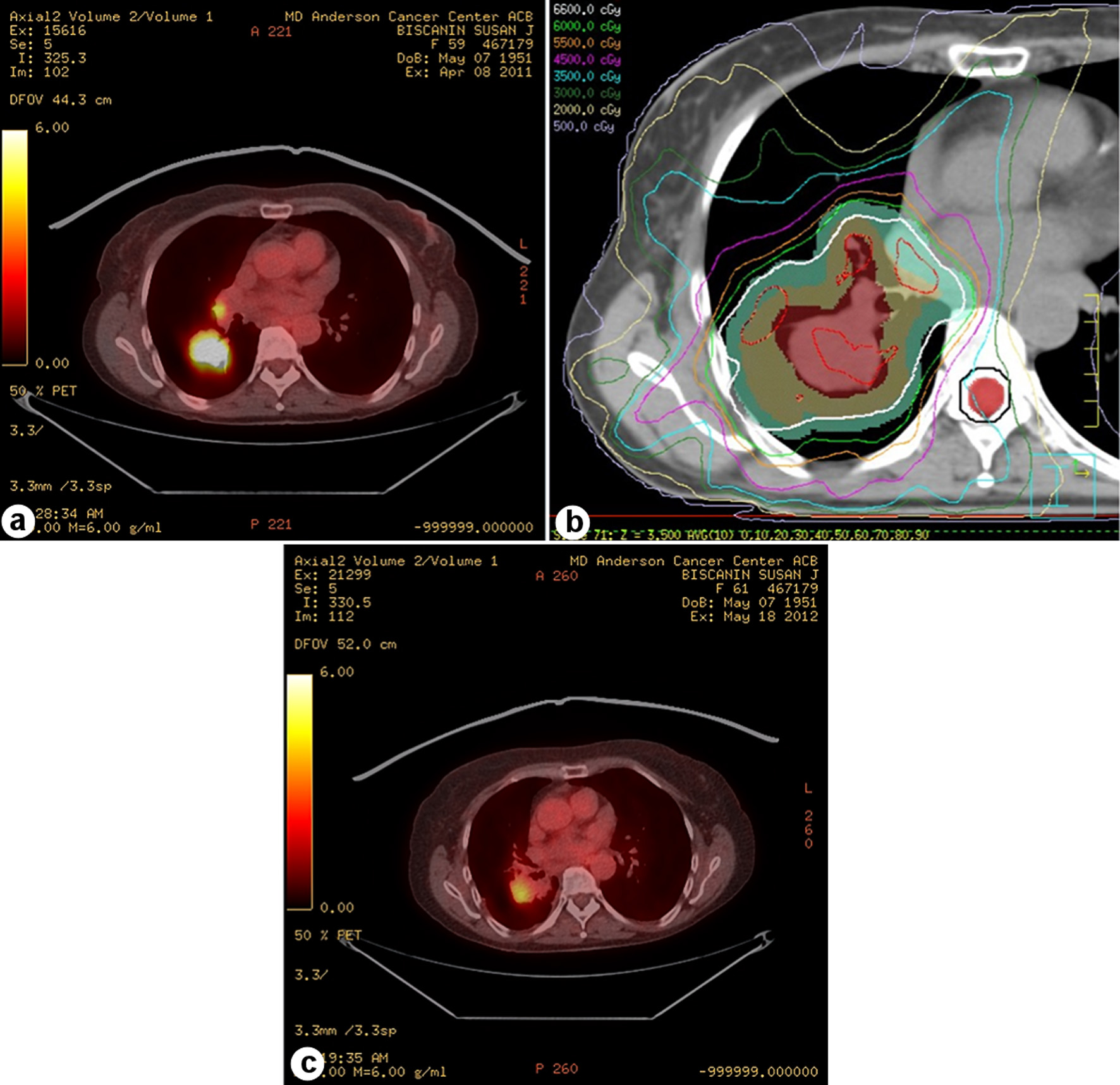 Click for large image | Figure 1. Local tumor progression in a patient with stage III adenocarcinoma lung cancer. (a) Positron emission tomography (PET) image from diagnosis. (b) Intensity-modulated photon radiation therapy plan with isodose lines and planning target volume (PTV) in green colorwash. (c) Post-treatment PET scan shows local tumor progression inside the PTV. |
Univariate and multivariate analyses to identify the potential predictors of local progression are shown in Tables 2 and 3, respectively. In the univariate analysis, only concurrent chemotherapy was associated with better LPFS (HR: 0.385; 95% CI: 0.182 - 0.815; P = 0.013) and having a larger GTV may have been linked with poorer LPFS (HR: 1.002; 95% CI: 0.999 - 1.004; P = 0.058). Neither EGFR nor KRAS status was associated with LPFS in the univariate analysis (Table 2, Fig. 2a, b). However, in multivariate analysis, after adjustment for concurrent chemotherapy, GTV, and mutation status, KRAS mutation was associated with poorer LPFS (KRAS mutation: HR: 1.961; 95% CI: 1.062 - 3.622; P = 0.031) (Table 3).
Click to view | Table 2. Univariate Analysis of Independent Predictors of Local Progression |
Click to view | Table 3. Multivariate Analysis of Independent Predictors of Local Progression |
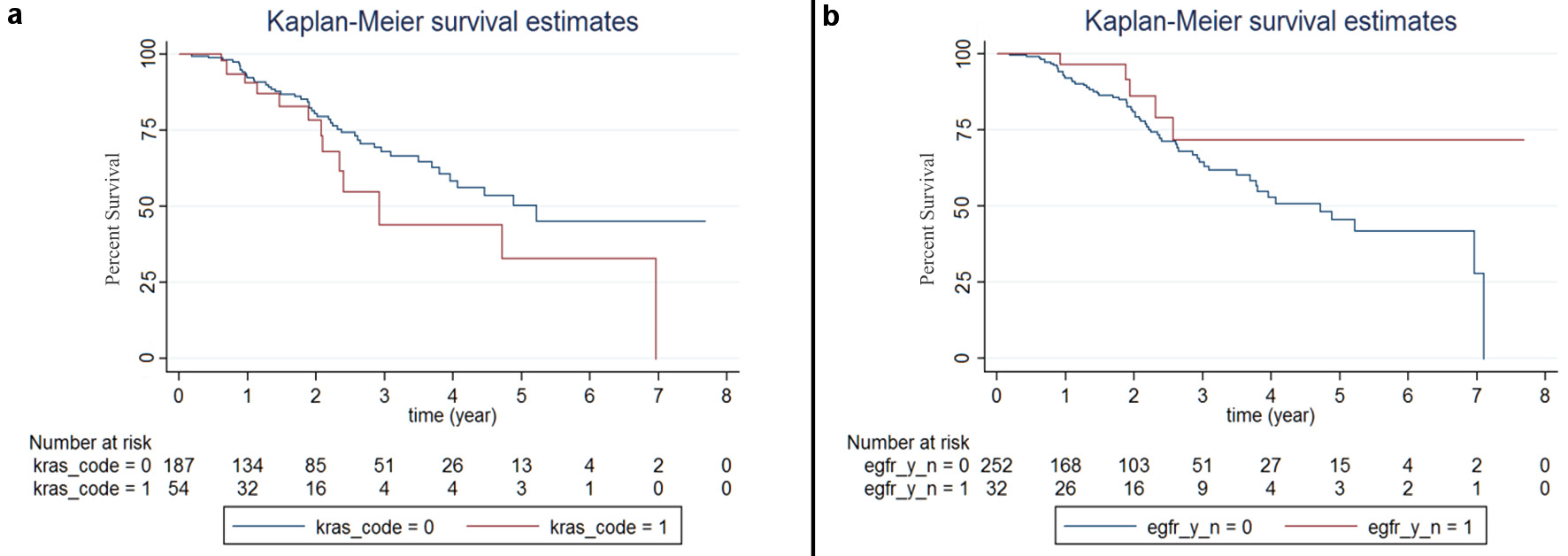 Click for large image | Figure 2. Kaplan-Meier plots of local progression-free survival (LPFS) according to EGFR and KRAS mutation status. (a) LPFS curves for patients with KRAS mutation (red line; krasm = 1), KRAS wild-type (WT; blue line (krasm = 0), P = 0.129). (b) LPFS curves for patients with EGFR mutation (red line; egfr = 1), EGFR WT (blue line; egfr = 0) P = 0.099. EGFR: epidermal growth factor receptor; KRAS: V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog. |
KRAS mutation is associated with radioresistance in human NSCLC cell lines
We hypothesized that NSCLC cell lines with KRAS mutation would be more resistant to radiation than cell lines with wild-type KRAS. To test this hypothesis, we performed clonogenic assays with two cell lines harboring KRAS mutations (H460 and A549) and two cell lines with wild-type KRAS (H1299 and H661). In support of our previous results in patients, we found that cell lines with KRAS mutations were more resistant to radiation than wild-type cells (Fig. 3a). In contrast, H1299 (KRAS wild-type (wt)) and H460 (KRAS mutation (mut)) were irradiated with ionizing radiation (4 Gy); immunofluorescence analyses were performed at the indicated time points. The number of γH2AX foci was then counted and quantified. Representative images of γH2AX foci in H460 (KRAS mut) and H1299 (KRAS WT) cells at each time point are shown in Figure 3b, c. The average (± SD) number of γH2AX foci per cell of control was not different between H460 and H1299 cell lines (7.61 ± 1.42 vs. 5.97 ± 1.13, P = 0.118). γH2AX foci were significantly higher after radiation, and significantly different from that at baseline at 1 h (25.32 ± 4.11 vs. 33.54 ± 1.93, P = 0.042) and 8 h (17.47 ± 3.51 vs. 28.3 ± 6.64, P = 0.043), and significantly higher at 24 h (12.33 ± 1.52 vs. 21.67 ± 3.78, P = 0.007) for the H1299 cell line (Fig. 3d).
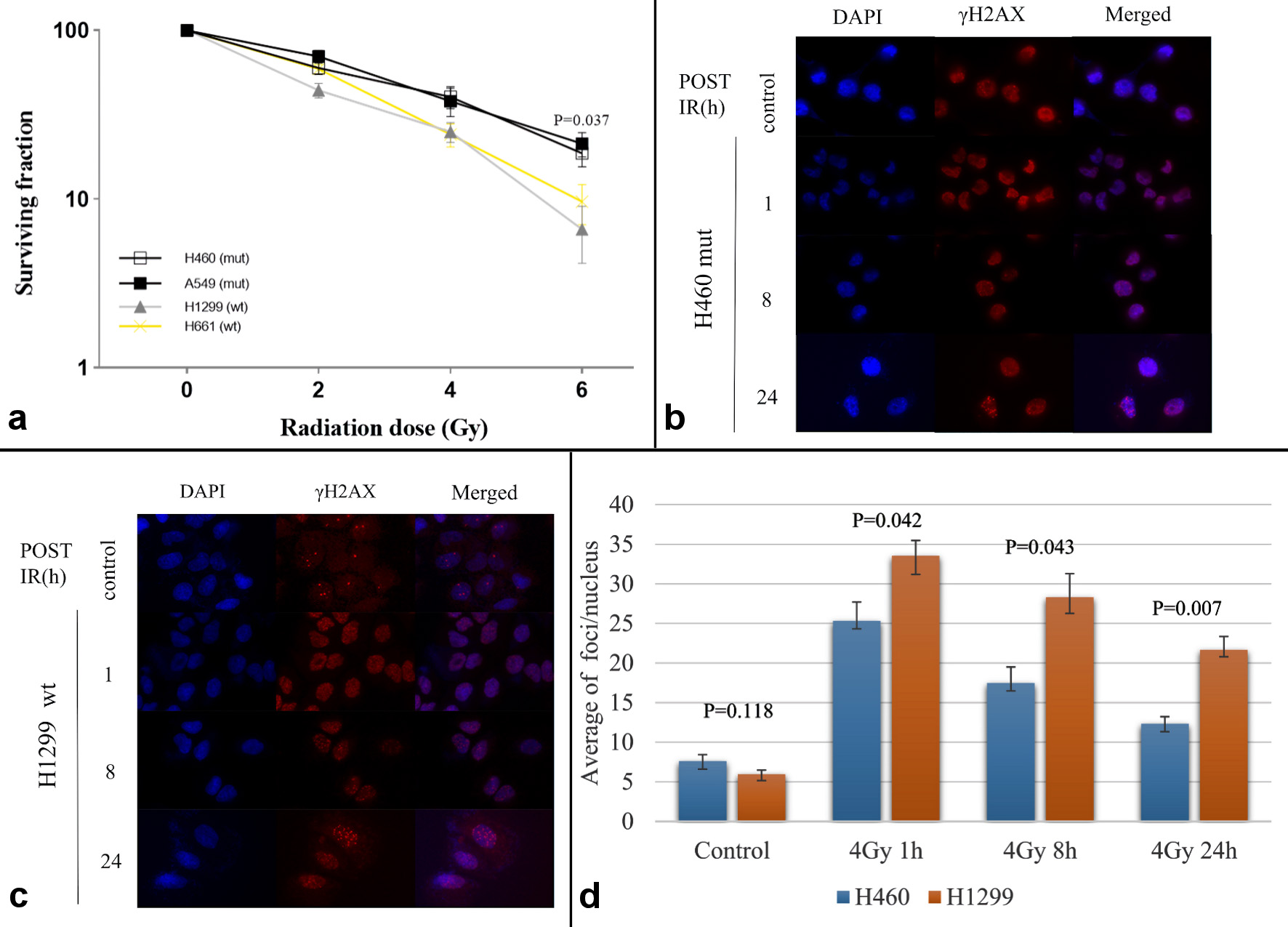 Click for large image | Figure 3. (a) Clonogenic assays showed that cell lines with KRAS mutation (H460 and A549) are more resistant than wild-type cells (H1299 and H661) to radiation. (b) H460 (KRAS mutation) and (c) H1299 (KRAS wild-type) treated with 4 Gy X-ray for 1, 8, and 24 h. Cells were then fixed and immune stained for γH2AX foci (red). Nuclei were counterstained with DAPI (blue). (d) For each time point, five to 10 images were captured and used for quantification of γH2AX foci number. The graph represents an average of three independent experiments ± SD. KRAS: V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog; DAPI: 4’,6-diamidino-2-phenylindole; SD: standard deviation. |
TCGA data showed multiple pathways involved in KRAS mutation patients and cell lines
TCGA LUAD data showed that the ERK1/2 pathway, phosphatidylinositol I3 kinase (PI3K)/mTOR, p38 MAPK pathway, cell cycle checkpoint signaling, DNA damage, repair pathways, and EGFR/PKC/AKT pathway were differentially expressed in KRAS mutations patients or cell lines relative to the wild-type group. Differentially expressed genes for patients with KRAS mutations are shown in Figure 4a. Raf-pS338, MEK1 pS217-S221, MAPK-pT202-Y204, and YB.1-Ps102 were upregulated, while ERK2 was downregulated in the ERK1/2 pathway. HER3, mTOR-pS2448, and S6-pS235-S236 were upregulated, and 4EBP1 was downregulated in the PI3K/mTOR pathway. p90RSK-pT359-S363 was upregulated, and PI3K.p110.alpha and STAT5.alpha were downregulated in the p38 MAPK pathway. Regarding cell cycle checkpoint signaling, X53BP1, Chk2, CyclinE1, and CyclinB1 were downregulated. PARP was upregulated and ATM, Ab.3, and PCNA were downregulated in DNA damage and repair signaling. For KRAS mutation cell, PKC.alpha, PKC.alpha pS657, PKC.delta pS664, IRS1, transglutaminase, MIG.6, Akt, and Y-box binding protein-1 (pS102) (YB-1 pS102) were upregulated, and EGFR pY1068, GAB2, and ShcpY317 were downregulated in the EGFR/PKC/AKT pathway (Fig. 4b). TCGA data from lung adenocarcinoma patients and cell lines showed that YB-1 pS102 was upregulated in the KRAS mutation group.
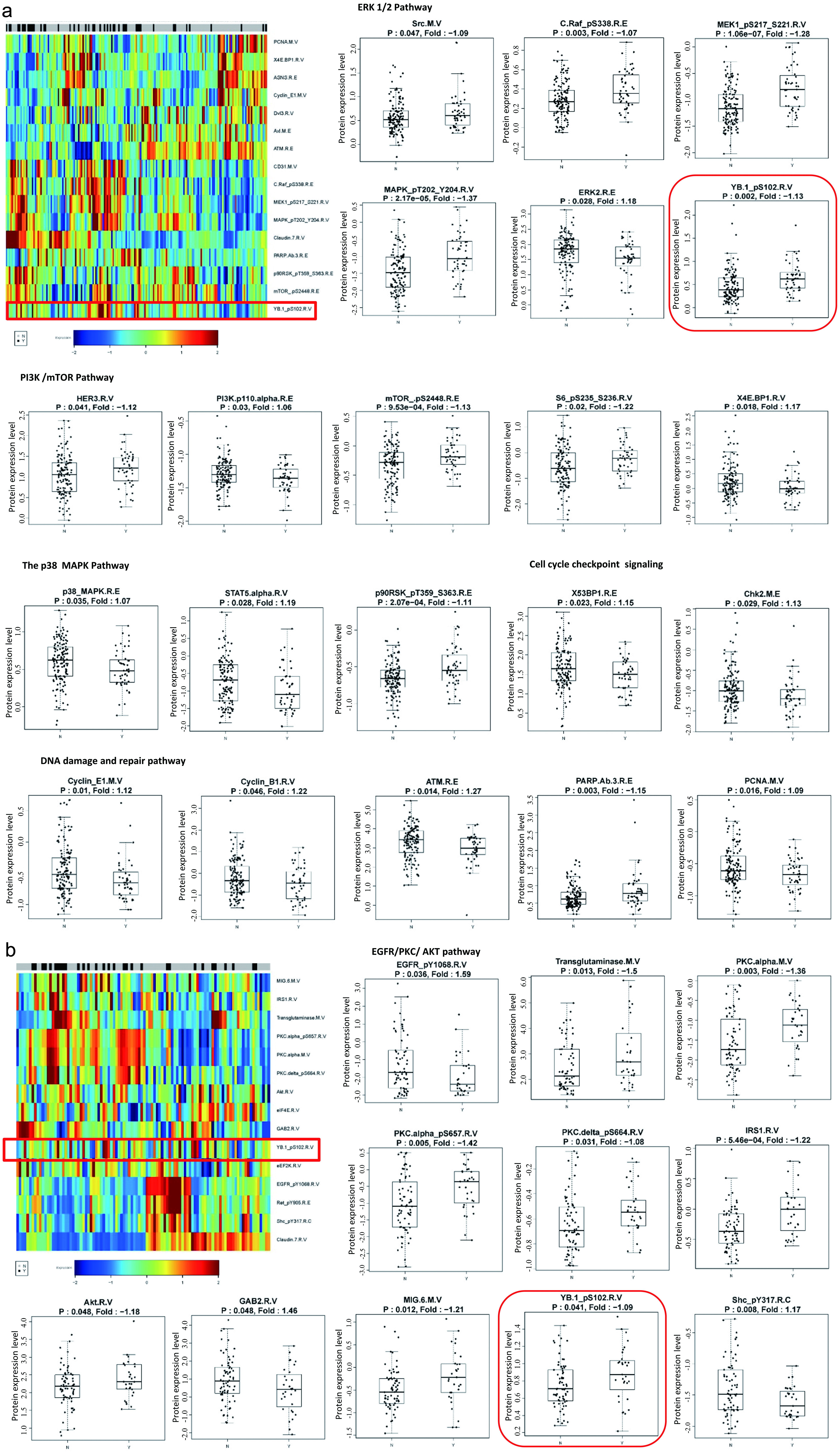 Click for large image | Figure 4. TCGA LUAD data show differentially expressed genes in the ERK1/2 pathway, phosphatidylinositol I3 kinase (PI3K)/mTOR, p38 MAPK pathway, cell cycle checkpoint signaling, and DNA damage. The repair and EGFR/PKC/AKT pathways presented with differential expression in patients (a) and cell lines (b) with KRAS mutations compared with the wild-type group. Y: patients or cell line with mutation; N: patients or cell line without mutation. TCGA LUAD: The Cancer Genome Atlas for Lung Adenocarcinoma. |
| Discussion | ▴Top |
In this study, we found that the presence of mutated KRAS in patients undergoing definitive radiation therapy for NSCLC was associated with inferior local control after adjustment for concurrent chemotherapy and GTV, suggesting that KRAS mutations may confer radioresistance in NSCLC. To the best of our knowledge, this is the first clinical study to show an association between KRAS mutation status and local tumor control in patients with NSCLC after definitive radiation therapy.
Local control is strongly linked with improved overall survival in locally advanced NSCLC [19, 21]. Assessment of local control after radiation therapy is governed by both the accuracy of detecting such diseases after treatment and the observation interval between treatment completion and progression or recurrence [22-24]. Hazuka et al [24] suggested that local progression can be diagnosed based on clinical, bronchoscopic, or radiographic evidence of tumor regrowth within the irradiated field. Martel et al [23] indicated that LPFS rates should be calculated at ≥ 30 months after radiation therapy. We also defined local progression according to PET, CT, and biopsy findings, and our median follow-up time for LPFS extended well beyond the recommended 30-month minimum (median, 57.4 months; range, 1.27 - 93.5 months).
Although others have found local control to be associated with performance status, concurrent chemotherapy, and radiotherapy dose [19], we found only one such association between local control and concurrent chemotherapy. Because the prescribed dose in our study was 68.7 ± 6.2 Gy (or Gy (RBE)), dose escalation did not improve local control [25], and the dose to the tumor field in our study met the requirement that 95% of the PTV received 100% of the prescription dose, we conclude that these instances of local tumor progression indicated intrinsic radioresistance.
Tumor radioresistance, whether inherent or acquired, is a significant obstacle in the effective treatment of NSCLC. The mechanisms influencing intrinsic radiosensitivity have suggested that up to 80% of this variability could have a genetic basis [26-28]. DNA double-strand breaks (DSBs) are thought to be the most severe molecular consequences of radiation therapy [29], and DSB repair is a determinant of cellular radiosensitivity [30]. Although several promising biomarkers of cellular radiosensitivity have been tested, there is insufficient evidence of their utility in clinical practice [31]. Therefore, we used the gold standard colony-survival assay to evaluate radiation sensitivity and the efficiency of DSB repair in NSCLC cell lines harboring different KRAS mutation status [32]. Another cellular radiosensitivity biomarker is γH2AX foci quantification [33]; H2AX is a central component of numerous signaling pathways in response to DSBs [34]. It is rapidly phosphorylated in response to DNA DSBs and contributes to repair protein recruitment to these damaged sites. H460 cells (KRAS mutation) showed lower induction of γH2AX a lower rate of foci disappearance after irradiation compared to H1299 cells (KRAS WT), which were considered to be correlated with radioresistance.
Our findings provide in vitro evidence that NSCLC cell lines transfected with a KRAS mutant are more resistant to radiation, consistent with the presence of a KRAS mutation associated with local control after definitive radiation therapy for NSCLC.
How does the mutation status of KRAS contribute to radio response? We compared the gene expression of different KRAS statuses in the TCGA LUAD database, which includes data on multiple pathways in KRAS signaling. Some studies have shown consistency with TCGA analysis [35-37]. After radiation, ligand-independent phosphorylation of EGFR can activate the RAS/RAF/MEK/MAPK, PI3K/AKT, and STAT3/STAT5 pathways; KRAS-mutated human tumor cell lines might activate EGFR via upregulated autocrine/paracrine production and secretion of EGFR ligands, resulting in an upregulation of the EGFR-PI3K-AKT-survival pathway [35], which is involved in the resistance of NSCLC to radiotherapy. It is considered responsible for the accelerated repopulation of tumor clonogenic cells during radiotherapy [38].
Interestingly, data from both the patient and cell lines showed that YB-1 pS102 was upregulated in the KRAS mutation group. YB-1 belongs to a family of DNA-binding proteins [39]. YB-1 is involved in many pathways, including the E2F pathway [40], PI3K/Akt kinase signaling [41], MAPK/ERK signaling [42], and EGFR pathways. It was also shown that radiation or mutated KRAS overexpression in breast cancer cell lines enhanced basal YB-1 phosphorylation and increased DNA DSB repair and post-irradiation survival [42]. Our future work will explore the role of YB-1 pS102 in KRAS mutation NSCLC.
Radiation therapy is the definitive treatment for NSCLC but is associated with high rates of local failure. KRAS is an essential predictor of the prognosis of NSCLC. However, its role in tumor response to radiation is not entirely clear. This study determined KRAS mutation status in conjunction with local tumor progression after definitive radiation therapy for NSCLC, indicating intrinsic radioresistance. Verified using a clonogenic assay and in vitro immunofluorescence, TCGA analysis was used to explore differential gene expression and potential pathways. Our findings could be helpful for the baseline prediction of outcomes according to KRAS genotype and may provide a potential target for radiosensitization in future studies.
Our conclusion from the current study was that KRAS mutations are associated with NSCLC. KRAS mutation status may be helpful as a biomarker of radioresistance and a potential target for increasing radiosensitivity.
Acknowledgments
The authors thank the patients and their families who made this study possible.
Financial Disclosure
This study was supported by the National Natural Science Foundation of China (grant numbers: 81673718 and 82074132) and Science and Technology Projects in Guangzhou (grant number: 202102080405).
Conflict of Interest
All authors have no conflict of interest to declare.
Informed Consent
Informed consents were obtained from all patients.
Author Contributions
Wei Xiao and Qin Fan designed this study. Dao Qi Zhu, Qin Fan, and Ai Wu Li coordinated the study and finalized the manuscript. Zhi Jian Yu, Ru Hua Zhang, and Feng Ying Gong performed the experiments. Dao Qi Zhu, Ying Liu, and Wei Wang analyzed the data. Ying Liu and Dao Qi Zhu wrote the paper.
Data Availability
Data supporting the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
WT: wild-type; HR: hazard ratio; CI: confidence interval; FDR: false discovery rate; GTV: gross tumor volume
| References | ▴Top |
- Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83(5):584-594.
doi - Curran WJ, Jr., Paulus R, Langer CJ, Komaki R, Lee JS, Hauser S, Movsas B, et al. Sequential vs. concurrent chemoradiation for stage III non-small cell lung cancer: randomized phase III trial RTOG 9410. J Natl Cancer Inst. 2011;103(19):1452-1460.
doi pubmed - Le Chevalier T, Arriagada R, Tarayre M, Lacombe-Terrier MJ, Laplanche A, Quoix E, Ruffie P, et al. Significant effect of adjuvant chemotherapy on survival in locally advanced non-small-cell lung carcinoma. J Natl Cancer Inst. 1992;84(1):58.
doi pubmed - Fournel P, Robinet G, Thomas P, Souquet PJ, Lena H, Vergnenegre A, Delhoume JY, et al. Randomized phase III trial of sequential chemoradiotherapy compared with concurrent chemoradiotherapy in locally advanced non-small-cell lung cancer: Groupe Lyon-Saint-Etienne d'Oncologie Thoracique-Groupe Francais de Pneumo-Cancerologie NPC 95-01 Study. J Clin Oncol. 2005;23(25):5910-5917.
doi pubmed - Furuse K, Fukuoka M, Kawahara M, Nishikawa H, Takada Y, Kudoh S, Katagami N, et al. Phase III study of concurrent versus sequential thoracic radiotherapy in combination with mitomycin, vindesine, and cisplatin in unresectable stage III non-small-cell lung cancer. J Clin Oncol. 1999;17(9):2692-2699.
doi pubmed - Zeng J, Aziz K, Chettiar ST, Aftab BT, Armour M, Gajula R, Gandhi N, et al. Hedgehog pathway inhibition radiosensitizes non-small cell lung cancers. Int J Radiat Oncol Biol Phys. 2013;86(1):143-149.
doi pubmed - Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39-85.
doi pubmed - Kerns SL, West CM, Andreassen CN, Barnett GC, Bentzen SM, Burnet NG, Dekker A, et al. Radiogenomics: the search for genetic predictors of radiotherapy response. Future Oncol. 2014;10(15):2391-2406.
doi pubmed - Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, Ince WL, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23(25):5900-5909.
doi pubmed - Sun JM, Hwang DW, Ahn JS, Ahn MJ, Park K. Prognostic and predictive value of KRAS mutations in advanced non-small cell lung cancer. PLoS One. 2013;8(5):e64816.
doi pubmed - Macerelli M, Caramella C, Faivre L, Besse B, Planchard D, Polo V, Ngo Camus M, et al. Does KRAS mutational status predict chemoresistance in advanced non-small cell lung cancer (NSCLC)? Lung Cancer. 2014;83(3):383-388.
doi pubmed - Mak RH, Hermann G, Lewis JH, Aerts HJ, Baldini EH, Chen AB, Colson YL, et al. Outcomes by tumor histology and KRAS mutation status after lung stereotactic body radiation therapy for early-stage non-small-cell lung cancer. Clin Lung Cancer. 2015;16(1):24-32.
doi pubmed - Johung KL, Yao X, Li F, Yu JB, Gettinger SN, Goldberg S, Decker RH, et al. A clinical model for identifying radiosensitive tumor genotypes in non-small cell lung cancer. Clin Cancer Res. 2013;19(19):5523-5532.
doi pubmed - Yagishita S, Horinouchi H, Sunami KS, Kanda S, Fujiwara Y, Nokihara H, Yamamoto N, et al. Impact of KRAS mutation on response and outcome of patients with stage III non-squamous non-small cell lung cancer. Cancer Sci. 2015;106(10):1402-1407.
doi pubmed - Minjgee M, Toulany M, Kehlbach R, Giehl K, Rodemann HP. K-RAS(V12) induces autocrine production of EGFR ligands and mediates radioresistance through EGFR-dependent Akt signaling and activation of DNA-PKcs. Int J Radiat Oncol Biol Phys. 2011;81(5):1506-1514.
doi pubmed - Wang M, Kern AM, Hulskotter M, Greninger P, Singh A, Pan Y, Chowdhury D, et al. EGFR-mediated chromatin condensation protects KRAS-mutant cancer cells against ionizing radiation. Cancer Res. 2014;74(10):2825-2834.
doi pubmed - Bernhard EJ, Stanbridge EJ, Gupta S, Gupta AK, Soto D, Bakanauskas VJ, Cerniglia GJ, et al. Direct evidence for the contribution of activated N-ras and K-ras oncogenes to increased intrinsic radiation resistance in human tumor cell lines. Cancer Res. 2000;60(23):6597-6600.
- Sun Y, Moretti L, Giacalone NJ, Schleicher S, Speirs CK, Carbone DP, Lu B. Inhibition of JAK2 signaling by TG101209 enhances radiotherapy in lung cancer models. J Thorac Oncol. 2011;6(4):699-706.
doi pubmed - Machtay M, Paulus R, Moughan J, Komaki R, Bradley JE, Choy H, Albain K, et al. Defining local-regional control and its importance in locally advanced non-small cell lung carcinoma. J Thorac Oncol. 2012;7(4):716-722.
doi pubmed - Rajpara RS, Schreibmann E, Fox T, Stapleford LJ, Beitler JJ, Curran WJ, Higgins KA. Locoregional tumor failure after definitive radiation for patients with stage III non-small cell lung cancer. Radiat Oncol. 2014;9:187.
doi pubmed - Auperin A, Le Pechoux C, Rolland E, Curran WJ, Furuse K, Fournel P, Belderbos J, et al. Meta-analysis of concomitant versus sequential radiochemotherapy in locally advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(13):2181-2190.
doi pubmed - Koto M, Miyamoto T, Yamamoto N, Nishimura H, Yamada S, Tsujii H. Local control and recurrence of stage I non-small cell lung cancer after carbon ion radiotherapy. Radiother Oncol. 2004;71(2):147-156.
doi pubmed - Martel MK, Ten Haken RK, Hazuka MB, Kessler ML, Strawderman M, Turrisi AT, Lawrence TS, et al. Estimation of tumor control probability model parameters from 3-D dose distributions of non-small cell lung cancer patients. Lung Cancer. 1999;24(1):31-37.
doi - Hazuka MB, Turrisi AT, 3rd, Lutz ST, Martel MK, Ten Haken RK, Strawderman M, Borema PL, et al. Results of high-dose thoracic irradiation incorporating beam's eye view display in non-small cell lung cancer: a retrospective multivariate analysis. Int J Radiat Oncol Biol Phys. 1993;27(2):273-284.
doi - Bradley JD, Paulus R, Komaki R, Masters G, Blumenschein G, Schild S, Bogart J, et al. Standard-dose versus high-dose conformal radiotherapy with concurrent and consolidation carboplatin plus paclitaxel with or without cetuximab for patients with stage IIIA or IIIB non-small-cell lung cancer (RTOG 0617): a randomised, two-by-two factorial phase 3 study. Lancet Oncol. 2015;16(2):187-199.
doi - Pao W, Kris MG, Iafrate AJ, Ladanyi M, Janne PA, Wistuba, II, Miake-Lye R, et al. Integration of molecular profiling into the lung cancer clinic. Clin Cancer Res. 2009;15(17):5317-5322.
doi pubmed - Das AK, Bell MH, Nirodi CS, Story MD, Minna JD. Radiogenomics predicting tumor responses to radiotherapy in lung cancer. Semin Radiat Oncol. 2010;20(3):149-155.
doi pubmed - Hornhardt S, Rossler U, Sauter W, Rosenberger A, Illig T, Bickeboller H, Wichmann HE, et al. Genetic factors in individual radiation sensitivity. DNA Repair (Amst). 2014;16:54-65.
doi pubmed - Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis. 2002;23(5):687-696.
doi pubmed - Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front Oncol. 2013;3:113.
doi pubmed - Chua ML, Rothkamm K. Biomarkers of radiation exposure: can they predict normal tissue radiosensitivity? Clin Oncol (R Coll Radiol). 2013;25(10):610-616.
doi pubmed - Nahas SA, Gatti RA. DNA double strand break repair defects, primary immunodeficiency disorders, and 'radiosensitivity'. Curr Opin Allergy Clin Immunol. 2009;9(6):510-516.
doi pubmed - Borras M, Armengol G, De Cabo M, Barquinero JF, Barrios L. Comparison of methods to quantify histone H2AX phosphorylation and its usefulness for prediction of radiosensitivity. Int J Radiat Biol. 2015;91(12):915-924.
doi pubmed - Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8(12):957-967.
doi pubmed - Toulany M, Dittmann K, Baumann M, Rodemann HP. Radiosensitization of Ras-mutated human tumor cells in vitro by the specific EGF receptor antagonist BIBX1382BS. Radiother Oncol. 2005;74(2):117-129.
doi pubmed - Toulany M, Baumann M, Rodemann HP. Stimulated PI3K-AKT signaling mediated through ligand or radiation-induced EGFR depends indirectly, but not directly, on constitutive K-Ras activity. Mol Cancer Res. 2007;5(8):863-872.
doi pubmed - Toulany M, Dittmann K, Kruger M, Baumann M, Rodemann HP. Radioresistance of K-Ras mutated human tumor cells is mediated through EGFR-dependent activation of PI3K-AKT pathway. Radiother Oncol. 2005;76(2):143-150.
doi pubmed - Milas L, Fan Z, Andratschke NH, Ang KK. Epidermal growth factor receptor and tumor response to radiation: in vivo preclinical studies. Int J Radiat Oncol Biol Phys. 2004;58(3):966-971.
doi pubmed - Evdokimova V, Tognon C, Ng T, Ruzanov P, Melnyk N, Fink D, Sorokin A, et al. Translational activation of snail1 and other developmentally regulated transcription factors by YB-1 promotes an epithelial-mesenchymal transition. Cancer Cell. 2009;15(5):402-415.
doi pubmed - Lasham A, Samuel W, Cao H, Patel R, Mehta R, Stern JL, Reid G, et al. YB-1, the E2F pathway, and regulation of tumor cell growth. J Natl Cancer Inst. 2012;104(2):133-146.
doi pubmed - Fujii T, Seki N, Namoto-Matsubayashi R, Takahashi H, Inoue Y, Toh U, Kage M, et al. YB-1 prevents apoptosis via the mTOR/STAT3 pathway in HER-2-overexpressing breast cancer cells. Future Oncol. 2009;5(2):153-156.
doi pubmed - Toulany M, Schickfluss TA, Eicheler W, Kehlbach R, Schittek B, Rodemann HP. Impact of oncogenic K-RAS on YB-1 phosphorylation induced by ionizing radiation. Breast Cancer Res. 2011;13(2):R28.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.