

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 14, Number 5, October 2023, pages 406-422

Development of a Machine Learning-Based Prognostic Model for Hormone Receptor-Positive Breast Cancer Using Nine-Gene Expression Signature
Takashi Takeshitaa, Hirotaka Iwasea, Rongrong Wub, Danya Ziazadehb, Li Yanc, Kazuaki Takabeb, d, e, f, g, i
aDepartment of Breast and Endocrine Surgery, Kumamoto City Hospital, Kumamoto, Japan
bBreast Surgery, Department of Surgical Oncology, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA
cDepartment of Biostatistics and Bioinformatics, Roswell Park Comprehensive Cancer Center, Buffalo, NY, USA
dDepartment of Surgery, University at Buffalo Jacobs School of Medicine and Biomedical Sciences, the State University of New York, Buffalo, NY, USA
eDepartment of Breast Surgery and Oncology, Tokyo Medical University, Tokyo, Japan
fDepartment of Surgery, Yokohama City University, Yokohama, Japan
gDepartment of Surgery, Niigata University Graduate School of Medical and Dental Sciences, Niigata, Japan
hDepartment of Breast Surgery, Fukushima Medical University, Fukushima, Japan
iCorresponding Author: Kazuaki Takabe, Breast Surgery, Department of Surgical Oncology, Roswell Park Comprehensive Cancer Center, Buffalo, NY 14263, USA
Manuscript submitted August 4, 2023, accepted August 28, 2023, published online September 20, 2023
Short title: Machine Learning-Based Prognostic Model for HR+ BC
doi: https://doi.org/10.14740/wjon1700
| Abstract | ▴Top |
Background: Determining the prognosis of hormone receptor positive (HR+) breast cancer (BC), which accounts for 80% of all BCs, is critical in improving survival outcomes. Stratifying individuals at high risk of BC-related mortality and improving prognosis has been the focus of research for over a decade. However, these tools are not universal as they are limited to clinical factors. We hypothesized that a new framework for predicting prognosis in HR+ BC patients can develop using artificial intelligence.
Methods: A total of 2,338 HR+ human epidermal growth factor receptor 2 negative (HER2-) BC cases were analyzed from Molecular Taxonomy of Breast Cancer International Consortium (METABRIC), The Cancer Genome Atlas (TCGA), and Gene Expression Omnibus (GEO) cohorts. Groups were then divided into high- and low-risk categories utilizing a recurrence prediction model (RPM). An RPM was created by extracting nine prognosis-related genes from over 18,000 genes using a logistic progression model.
Results: Risk classification by RPM was significantly stratified in both the discovery cohort and validation cohort. In the time-dependent area under the curve analysis, there was some variation depending on the cohort, but accuracy was found to decline significantly after about 10 years. Cell cycle related gene sets, MYC, and PI3K-AKT-mTOR signaling were enriched in high-risk tumors by the Gene Set Enrichment Analysis. High-risk tumors were associated with high levels of immune cells from the lymphoid and myeloid lineage and immune cytolytic activity, as well as low levels of stem cells and stromal cells. High-risk tumors were also associated with poor therapeutic effects of chemotherapy and endocrine therapy.
Conclusions: This model was able to stratify prognosis in multiple cohorts. This is because the model reflects major BC therapeutic target pathways and tumor immune microenvironment and, further is supported by the therapeutic effect of chemotherapy and endocrine therapy.
Keywords: Breast cancer; Recurrence prediction; Cancer genomics; Tumor immune microenvironment; Machine learning
| Introduction | ▴Top |
Breast cancer (BC) is currently one of the most common types of cancer in women, and is increasing in incidence by 0.5% per year [1]. Approaches to improve the prognosis of hormone receptor positive (HR+) BC, which accounts for 80% of all BCs, is vital to enhancing BC survival. Improving stratification of individuals at high risk of BC-related mortality has been the focus of research interest for over a decade [2]. Researchers have mainly classified BC patients by different gene expression profiles and stratified according to clinical outcome [3, 4]. Such studies have evolved into the development of algorithms for estimating the risk of recurrence and survival by molecular gene expression signatures [5]. Some of these molecular prognostic indicators have since been recommended by the guidelines of ASCO and are now available for the clinical management of BC.
Another approach to improve the outcome of HR+ BC is identification of predictive biomarkers. For HR+ human epidermal growth factor receptor 2 negative (HER2-) early-stage BC, endocrine therapy (ET) is the main treatment, and the indication for adjuvant chemotherapy (CT), which has serious side effects, should be determined on a patient-by-patient basis. Therefore, additional prognostic information is often needed to provide patients with reliable and effective treatment. Five prognostic signatures for BC (OncotypeDX®, MammaPrint®, Prosigna®, EndoPredict®, and Breast Cancer IndexSM) which are included in national and international guidelines (NCCN, ASCO, ESMO, NICE, AGO, and St. Gallen) are representative and have been summarized in a recent review [5]. In particular, Oncotype DX® has been successful in focusing on the additional benefits of CT for HR+ BC [6-8].
However, these tools are not universal, as they are limited to clinical factors such as HRs, menopausal status, and nodular status [9]. It remains unclear which gene assay should be prioritized, and prognosis and prediction differ between tests [5]. There remains a significant need for an unbiased and comprehensive approach to identify and list all prognosis-related molecules [5, 10-12]. Innovations in high-throughput technology have led to the rapid accumulation of data on gene expression throughout the transcriptome of tumors from large numbers of patients. The study of such accumulated data by the research community is a very important study to identify more accurate potential biomarkers at the individual patient level [13]. With these cohorts and algorithms, we have conducted studies that assess the real-world relevance of expression of genes of interest [14-24].
We aimed to test the hypothesis that artificial intelligence can be used to develop a new framework for predicting prognosis in BC patients in multiple validation cohorts.
| Materials and Methods | ▴Top |
In all cohorts, given that the patient data are de-identified, and that it is in a public domain, it waived Institutional Review Board approval.
Key resources are shown in Table 1 [24-39].
 Click to view | Table 1. Key resources |
Study design and cohorts
We performed a retrospective analysis of six independent HR+ HER2- BC cohorts, which included 1,355 women from the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) cohort, 585 women from The Cancer Genome Atlas (TCGA) cohort, and a total of 398 women from the Gene Expression Omnibus (GEO) cohorts (GSE199135, GSE9195, GSE6532, and GSE21653) in which transcriptomic data were associated with clinical parameters, all of which were previously published [14-24, 40]. We downloaded public data from cBioPortal [41, 42] with the CGDS-R package for METABRIC (METABRIC Nature 2012 and Nat Commun 2016 dataset) and TCGA (TCGA PanCancer Atlas dataset) and the GEOquery package for GEO cohorts. We added clinical data from Rueda et al, 2019 dataset for METABRIC [43], Liu et al, 2018 dataset for TCGA [44], Takeshita et al, 2022 dataset for GSE199135 [24], Loi et al, 2010 dataset for GSE9195 and GSE6532 [25], and Sabatier et al, 2011 dataset for GSE [26] to each cohort (Supplementary Material 1, www.wjon.org).
The initial analysis was conducted with the METABRIC cohort (discovery cohort), given that this is the best-characterized cohort available. We then performed gene analysis differentially expressed with |log2 fold-change| greater than 0.25 and adjusted P < 0.05 in distant recurrence and identified 155 genes (Supplementary Material 2, www.wjon.org). For the 155 extracted human protein-coding genes, we used the Cox proportional-hazards (Cox-PH) model to examine the potential utility of each gene as a prognostic marker. Prognostic-related genes were defined as genes with P-values less than 0.05 in the Cox-PH model. Consequently, we uncovered 23 genes closely involved in distant recurrence free survival (RFS) in METABRIC HR+ HER2- patients (Supplementary Material 3, www.wjon.org). Furthermore, we combined the Cox-PH model with recursive feature elimination to narrow down the nine best predictors (Table 2). After validating several artificial intelligence-based machine learning algorithms, we used a logistic regression model (LRM) to optimize the weights of the nine selected genes and build an RPM. This model was generated using the Python-based scikit-learn library. To explore whether this model can stratify prognosis, we analyzed the METABRIC total recurrence, local recurrence cohort, TCGA BC cohort, and another independent BC cohort, GSE199135, GSE9195, GSE6532, and GSE21653 to verify its performance.
Click to view | Table 2. The Nine Genes Best Predictors Extracted From 23 Signature Genes Using the Cox-PH Model With Recursive Feature Elimination |
Cluster analysis
We chose hierarchical clustering using the Euclidean distance and Ward’s linkage due to its relative good performance [45]. The R-function “hclust” was used for performing hierarchical clustering.
Gene Set Enrichment Analysis (GSEA)
GSEA was performed comparing high and low risk of recurrence in RPM among hallmark gene sets using software provided by the Broad Institute [46], as we described previously [14, 15, 17]. We only considered gene sets significantly enriched that met a threshold of normalized enrichment score (NES) > 1.6 or < -1.6 and false discovery rate (FDR) q-value < 0.025. Gene Set Variation Analysis (GSVA) from the MSigDB Hallmark collection [47] was used to score the cancer hallmark gene sets for analysis. In doing so, we used the GSVA Bioconductor package (version 3.10), as we described previously [24].
Tumor microenvironment (TME) analysis
xCell, which is the bioinformatics tool that performs cell type enrichment analysis from gene expression data for 64 immune and stroma cell types, was used for TME analysis [48], as we described previously [24]. The immune cytolytic activity was defined as the geometric mean of GZMA and PRF1 expression values in Transcripts Per Million [49, 50] and immune cytolytic activity was calculated as previously described [16-24].
Statistical analysis
All statistical analyses were performed using R software [39] and Bioconductor [51] and Python (version 3.10.7 [33]). The Chi-square test or Fisher’s exact test or the nonparametric Mann-Whitney U test and contingency analysis were used to assess baseline differences between binary variables. In the analysis of RFS, the Kaplan-Meier method was used to estimate survival rates, and differences between survival curves were evaluated by the log-rank test. Two-sided P-value < 0.05 was considered as statistically significant for all tests.
| Results | ▴Top |
Extraction of all prognosis-related genes and preparation of an RPM using machine learning
In order to build an RPM, we examined the relation between abundance of mRNA expression and distant recurrence with METABRIC as a discovery cohort. Figure 1a shows volcano plots that represent the distribution of the fold changes and adjusted P-values of 18,484 genes in METABRIC HR+ HER2- cohort with and without distant recurrence. We identified 155 distant recurrence-related genes, which were differentially expressed with |log2 fold-change| greater than 0.25 and adjusted P < 0.05, with the complete list of these genes and their adjusted P values being provided in Supplementary Material 2 (www.wjon.org). A bidirectional hierarchical clustering heatmap, based on the expression levels of the identified differentially expressed genes (DEGs), indicated the relationship between clustering of the samples into two groups and type of recurrence (Fig. 1b). Analysis of the combined hazard ratio logarithms of all 155 extracted genes suggests that further refinement is necessary for prognostic prediction (Fig. 1c). Using the Cox-PH model, the optimal combination of prognostic genes was further screened from the 155 feature genes. Consequently, 23 DEGs were revealed to be closely involved in METABRIC HR+ HER2- patients’ distant RFS (P < 0.05) (Supplementary Material 3, www.wjon.org). Combining the Cox-PH model and recursive feature elimination, we sequentially removed genes with P ≥ 0.05 from the 23 genes, and finally extracted the nine best predictors with P < 0.05 (Table 2).
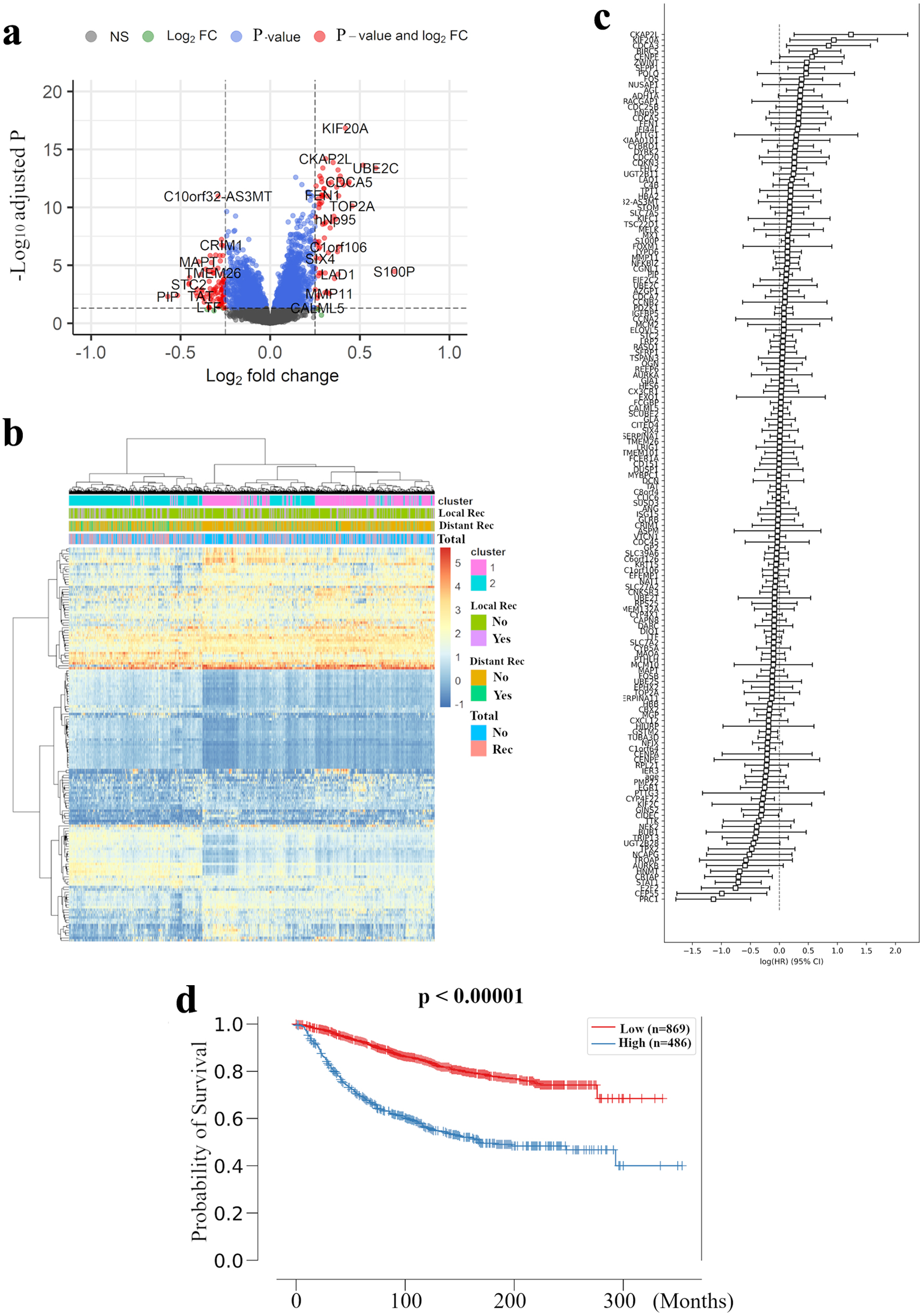 Click for large image | Figure 1. Extraction of all prognosis-related genes and preparation of a recurrence prediction model using machine learning. (a) Volcano plot illustrating the differentially expressed mRNAs of BC patients comparing with and without distant recurrence in the METABRIC HR+ HER2- cohort are shown. X-axes: log2 FC; Y-axes: -log 10 adjusted P-value from limma analysis. mRNAs with adjusted P-value < 0.05 and log2 FC > 0.25 are marked in red, with adjusted P-value > 0.05 and log2 FC < 0.25 in green, with adjusted P-value < 0.05 and log2 FC < 0.25 in blue, all others in black. (b) A heatmap illustrating the expression intensity of 155 genes extracted by (a), with colors ranging from red to blue as indicated in the key are shown. Both rows and columns are clustered using correlation distance and average linkage. (c) Logarithm of the integrated hazard ratio for all 155 genes extracted by (a) are shown. The complete list of these genes identified by meta-analysis is provided in Supplementary Material 1 (www.wjon.org). (d) Kaplan-Meier curves for distant RFS in METABRIC HR+ HER2- patients based on high and low risk in recurrence prediction model are shown. BC: breast cancer; METABRIC: Molecular Taxonomy of Breast Cancer International Consortium; FC: fold change; RFS: recurrence free survival; HR+: hormone receptor positive; HER2: human epidermal growth receptor 2; LRM: logistic regression model. |
In these nine best predictors, KIF20A and PRC1 were the most promising prognosis-related genes, with the highest and lowest hazard ratios, respectively. It is of note that one of these two genes, KIF20A, was not adopted by the MammaPrint or Oncotype Dx 21-gene RS systems. Indeed, most of the validated prognosis-related genes have not been well characterized to date with regard to their relation to basic or clinical oncology, with CYP4F22, TUBA3D, and HNMT receiving even less study.
Further, we investigated whether these nine newly identified prognosis-related genes were sufficient to predict survival in BC patients. An LRM was used to optimize the weights of the nine selected genes to build an RPM. In the METABRIC HR+ HER2- cohort as the training cohort, the model was set up to significantly discriminate the high risk of recurrence group with the log-rank P value of < 0.00001 for the Kaplan-Meier survival curve (Fig. 1d).
Validation of RPM
To validate whether this RPM can universally stratify prognosis, we examined TCGA HR+ HER2- BC cohort and other independent HR+ HER2- BC cohorts, GSE199135, GSE9195, GSE6532, and GSE21653, using the Kaplan-Meier method and verified by the log-rank test. The Kaplan-Meier survival curve shows that the log-rank P-values for RFS time in the validation sets were all < 0.05 (Fig. 2), suggesting significantly different RFS time between predicted recurrence and non-recurrence samples. A time-dependent area under the receiver operating characteristics curve (AUC) value was created to examine the details of each cohort’s timely accuracy (Supplementary Material 4, www.wjon.org). The AUC values peaked above 0.8 at GSE199135, GSE6532, and GSE21653. GSE199135 and GSE6532 maintained accuracy for about 10 years, but GSE21653 decreased in accuracy after 5 years. In METABRIC, TCGA, and GSE9195, the peak had not exceeded 0.8, but accuracy was also maintained for about 10 years. These results suggest that the RPM can universally and predominantly stratify prognosis in all HR+ HER2- BC cohorts. In the time-dependent AUC analysis, there was some variation depending on the cohort, but accuracy tended to decline significantly after about 10 years.
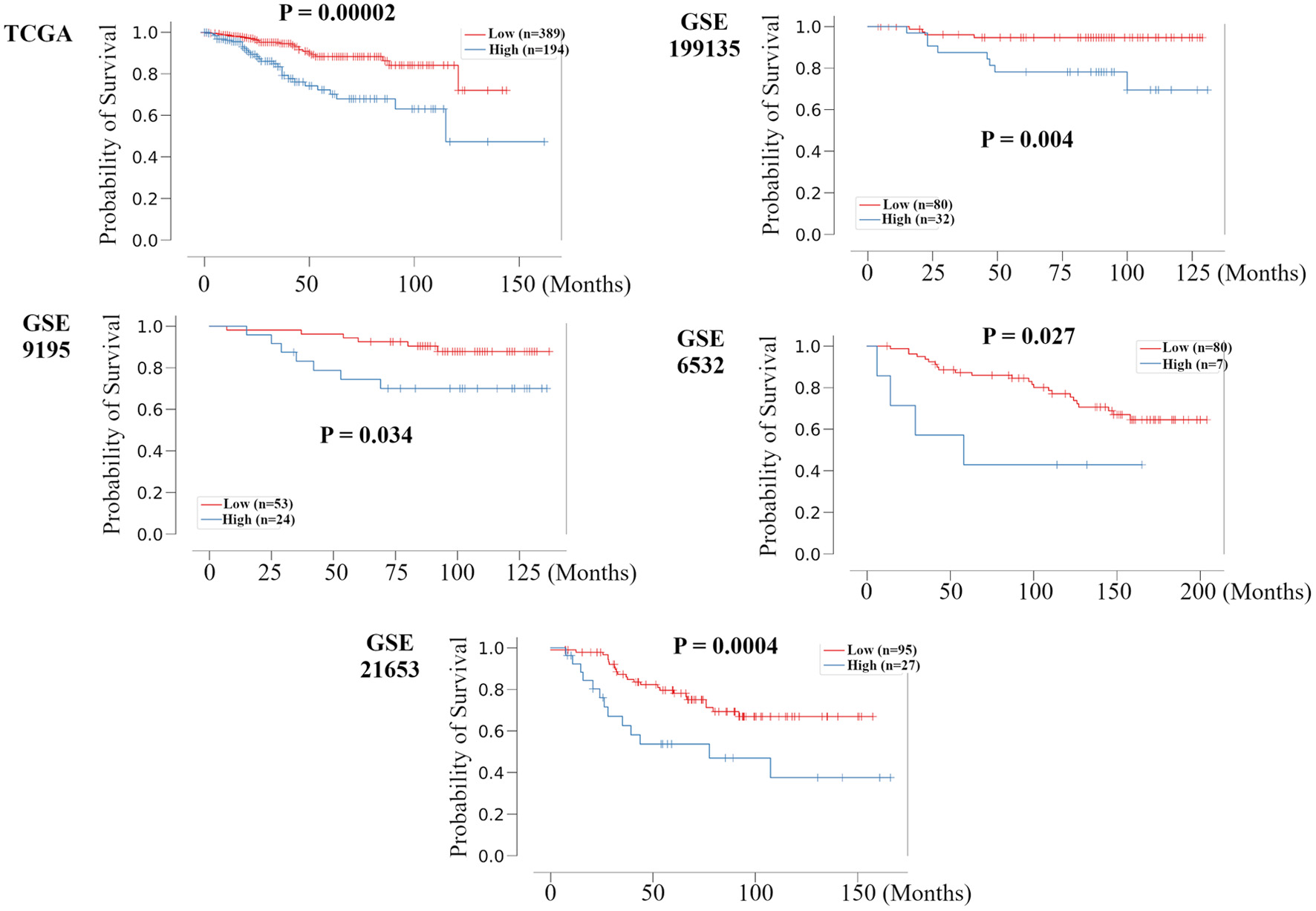 Click for large image | Figure 2. Validation of the relationship between the recurrence prediction model and survival rate in other HR+ HER2- BC cohorts. Kaplan-Meier plots of the association of the recurrence prediction model with RFS for the recurrence prediction model, applied on the TCGA, GSE199135, GSE9195, GSE6532, and GSE21653 are shown. HR+: hormone receptor positive; HER2: human epidermal growth receptor 2; BC: breast cancer; RFS: recurrence free survival; TCGA: The Cancer Genome Atlas; FC: fold change. |
High-risk tumors were associated with more aggressive clinical phenotype
Next, we used various factors to justify the risk classification by RPM as follows.
We studied the relationship between clinical features of the primary tumor and high and low risk in RPM amongst the HR+ HER2- subgroup in the METABRIC cohort (Table 3). Patients categorized as high risk in the RPM were significantly associated with increased age, postmenopausal state, higher tumor size, lymph node metastasis, invasive ductal carcinoma, higher grade, progesterone receptor (PgR) positivity, luminal B subtype, HER2 subtype, and basal-like subtype.
Click to view | Table 3. Patients and Clinical Characteristics Associated With Recurrence Prediction Model in METABRIC HR+ HER2- Cohort |
Cell cycle related gene sets, MYC, and PI3K-AKT-mammalian target of rapamycin (mTOR) signaling were enriched in high-risk tumors in the GSEA
In order to investigate which mechanism is related to the RPM, we examined gene sets associated with high-risk tumors in the RPM using the GSEA (Fig. 3). Pathway enrichment analysis revealed that five significant pathways were enriched for high-risk tumors; cell cycle related gene sets (mitotic spindle; NES = 1.98, FDR q < 0.0001, G2/M check point; NES = 1.83, FDR q = 0.003, E2F targets; NES = 1.76, FDR q = 0.006), MYC target v2 (NES = 1.68, FDR q = 0.006), and PI3K-AKT-mTOR signaling (NES = 1.68, FDR q = 0.012). No gene set was enriched among 50 hallmark gene sets with low-risk tumors.
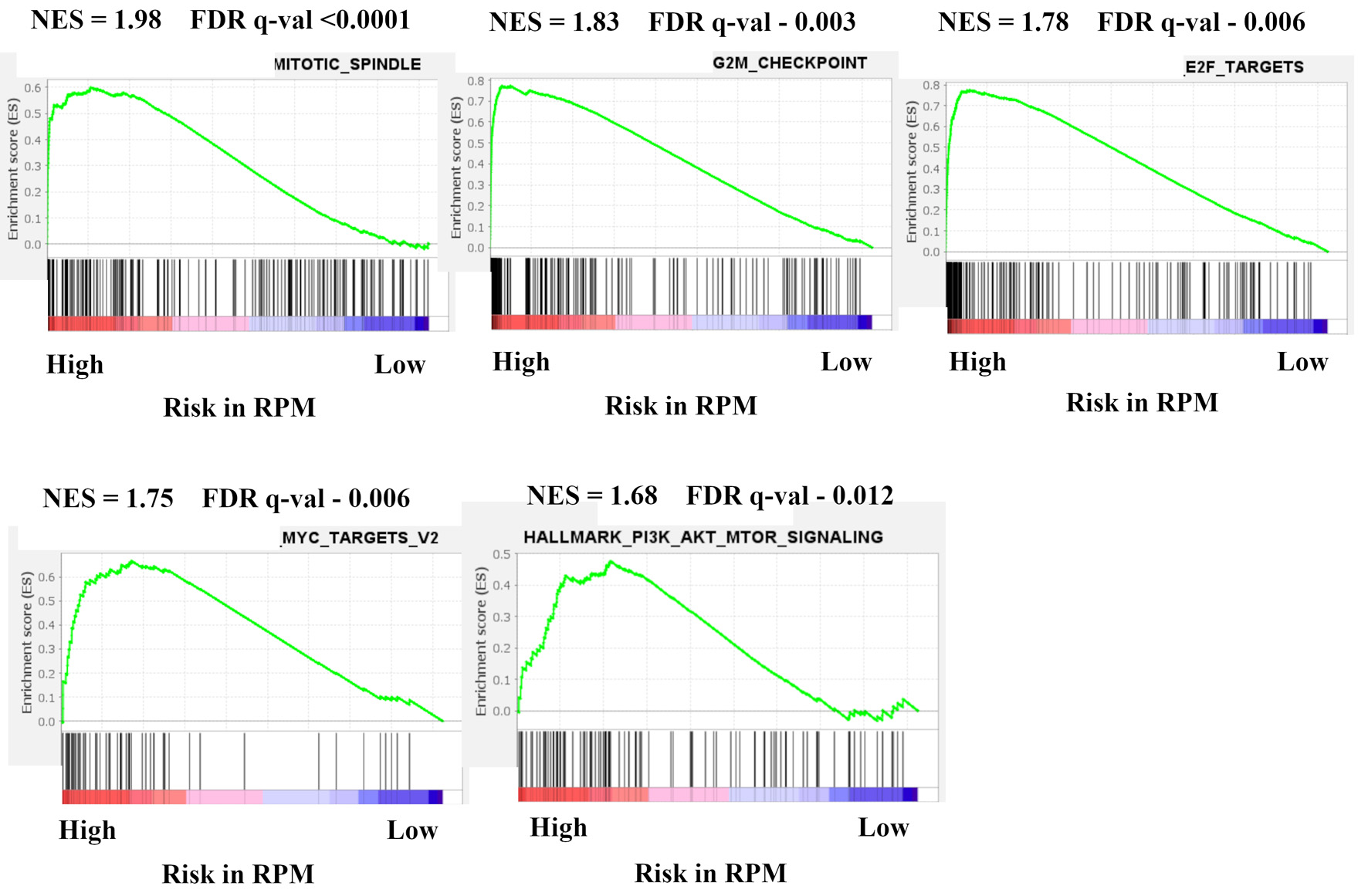 Click for large image | Figure 3. Gene expression profiles based on high and low risk in recurrence prediction model. GSEA of BC patients in METABRIC HR+ HER2- cohort comparing high and low risk in recurrence prediction model are shown. Upregulated pathways included mitotic spindle, G2/M check point, E2F targets, MYC target v2, and PI3K-AKT-mTOR signaling in high risk compared with low risk in LRM. The significance of each pathway was classified by a threshold of NES > 1.6 or < -1.6 and FDR q-value < 0.025. GSEA: Gene Set Enrichment Analysis; BC: breast cancer; METABRIC: Molecular Taxonomy of Breast Cancer International Consortium; HR+: hormone receptor positive; HER2: human epidermal growth receptor 2; mTOR: mammalian target of rapamycin; LRM: logistic regression model; NES: normalized enrichment score; FDR: false discovery rate. |
High-risk tumors were associated with high levels of immune cells from the lymphoid and myeloid lineage and immune cytolytic activity, and low levels of stem cells and stromal cells
We explored the difference in TME composition among high- and low-risk tumors in the RPM utilizing xCell (Fig. 4). High-risk tumors had a higher fraction of immune cells, and a decreased fraction of stem cells and stromal cells. For innate immune cells, eosinophils and macrophages were more prevalent in high-risk tumors, whereas mast cells and monocytes were lower. There was no significant difference in dendritic cells between high- and low-risk tumors in RPM. However, immature dendritic cells and conventional dendritic cells were significantly lower, whereas activated dendritic cells were significantly higher in the high-risk group. CD4+ naive T cells, CD4+ T cells, central memory CD4+ T cells, effector memory CD8+ T cells, class-switched memory B cells, naive B cells, natural killer T cells, pro B cells, Tgd, Th1, Th2 cells, and regulatory T cells (Tregs) were also significantly higher in the high-risk tumor group, but effector CD4+ T cells, central memory CD8+ T cells, and plasma cells were found to be significantly lower. The immune cytolytic activity score has been well established measure of the overall cytolytic activity of immune effector cells in bulk tumors [49]. We found that the immune cytolytic activity scores in BC tumors were significantly higher in the high-risk group. Additionally, in stromal cells, we found that representative stromal cells, such as endothelial cells, were significantly lower amongst the high-risk group. The xCell package enabled the generation of the stroma score using the sums of fractions of certain cell types [48]. We found that stroma scores in BC tumors were significantly lower in the high-risk group. These results indicate that high-risk patients in the RPM had a higher fraction and activity of immune cells, whilst simultaneously having a lower fraction of stem cells and stromal cells in breast TME.
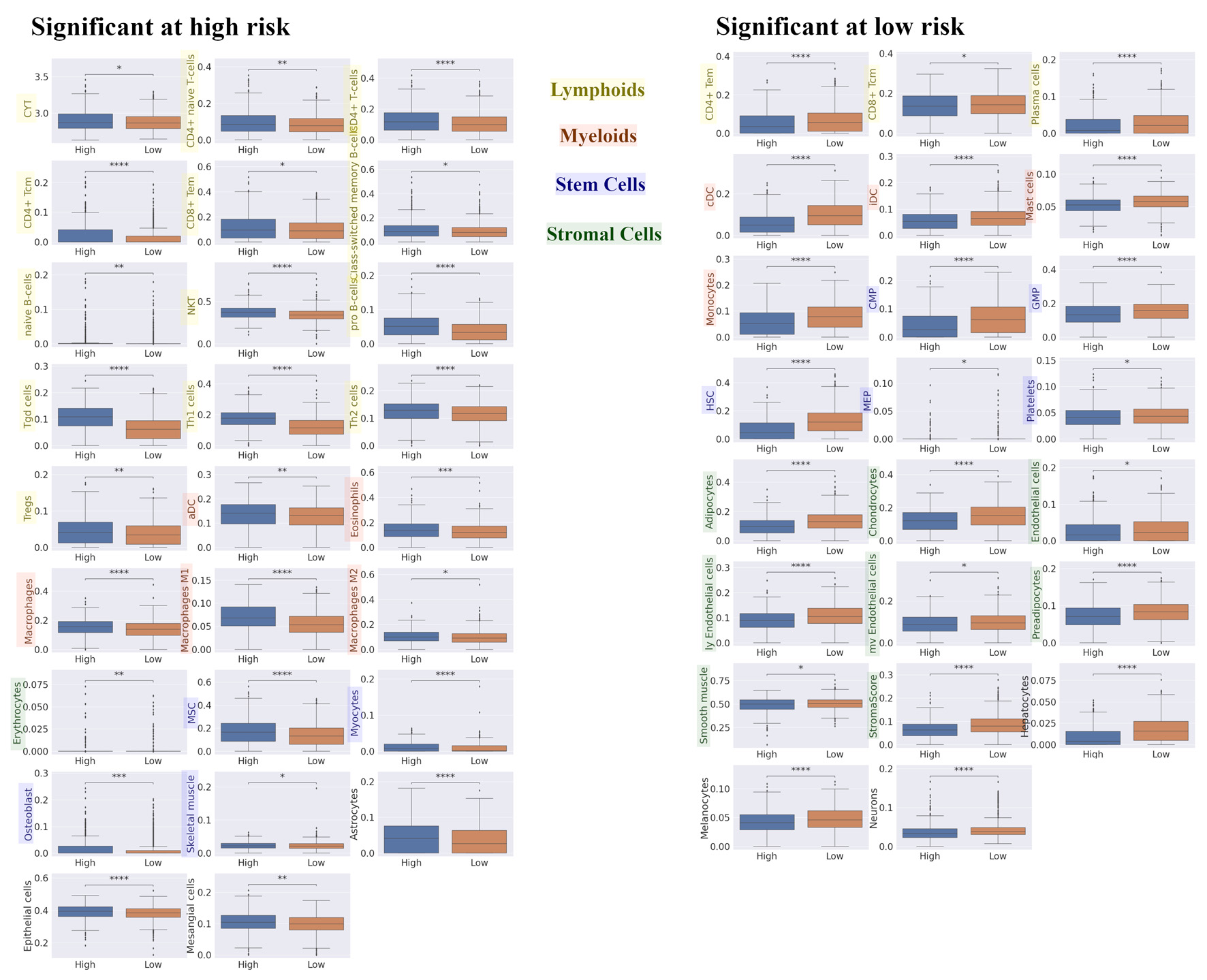 Click for large image | Figure 4. Differences in TME compositions for high and low risk in recurrence prediction model. We explored the difference in TME composition between high- and low- risk in recurrence prediction model utilizing xCell. Box plot of the relationship between recurrence risk in recurrence prediction model and TME in METABRIC HR+ HER2- cohort are shown. The left panel shows the cell fraction with up-regulation in high risk, and the right panel shows the cell fraction with up-regulation in low risk. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. TME: tumor microenvironment; METABRIC: Molecular Taxonomy of Breast Cancer International Consortium; HR+: hormone receptor positive; HER2: human epidermal growth receptor 2; CYT: immune cytolytic activity; CD4+ tcm: the central memory CD4+ T cell; CD8+ tem: the effector memory CD8+ T cell; NKT: natural killer T cells; Tregs: regulatory T cells; aDC: activated dendritic cell; MSC: mesenchymal stem cell; CD4+ tem: the effector memory CD4+ T cell; CD8+ tcm: the central memory CD8+ T cell; cDC: conventional dendritic cell; iDC: immature dendritic cell; CMP: common myeloid progenitor; GMP: granulocyte-macrophage progenitor; HSC: hematopoietic stem cell; MEP: megakaryocyte-erythroid progenitor. |
High-risk tumors among patients in the RPM were associated with poor treatment efficacy, which was supported by their correlations with pathways and tumor immune microenvironment (TIME) involved in treatment resistance
Here, we examined the relationship between risk classification by RPM and treatment prognosis for each recurrence type. The treatment options were ET and CT, which were originally noted in the METABRIC HR+ HER2- dataset. In distant recurrence and total recurrence analysis, patients in high-risk group were associated with poor prognosis in all treatment groups (Fig. 5). However, in local recurrence analysis, patients in high-risk group were associated with poor prognosis only in the ET group. These results suggest that risk classification by RPM was associated with the effect of treatment, especially the effect of ET in HR+ HER2- BC patients.
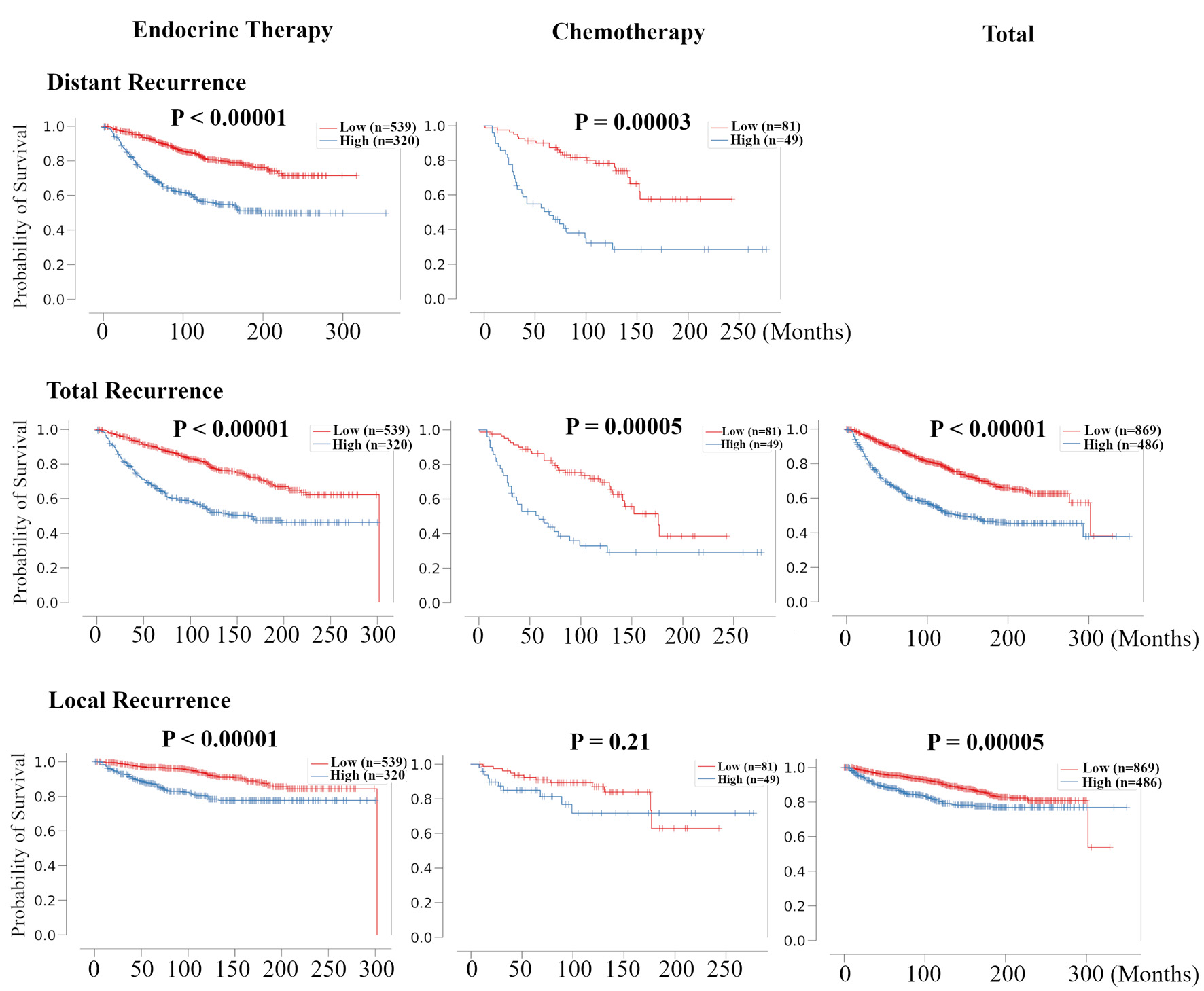 Click for large image | Figure 5. Validation of the relationship between risk classification in recurrence prediction model and the therapeutic effect of chemotherapy and endocrine therapy for HR+ HER2- BC patients. Kaplan-Meier plots of distant RFS, total RFS, and local RFS of the association between recurrence risk in recurrence prediction model and chemotherapy- and endocrine-treated patients in METABRIC HR+ HER2- cohort are shown. For total RFS and local RFS, Kaplan-Meier plots of total recurrence are shown on the far right. HR+: hormone receptor positive; HER2: human epidermal growth receptor 2; RFS: recurrence free survival; METABRIC: Molecular Taxonomy of Breast Cancer International Consortium. |
Further, we explored the relationship between treatment outcomes and signaling pathways using GSVA, immune cytolytic activity, and immune cell composition in the METABRIC HR+ HER2- cohort (Fig. 6). Based on treatments and recurrence, we classified patients into following three groups: patients treated with CT but relapsed as “CT rec”, patients treated with ET alone but relapsed as “ET rec”, and patients treated with ET with or without CT and who did not relapse as “No rec”. E2F targets, G2M checkpoint, Myc targets v2, and PI3K-AKT-mTOR signaling GSVA scores were highest in the “CT rec” group and lowest in the “No rec” group. High risk of RPM tumors was indeed correlated with these signaling pathways (Fig. 3). Other notable findings were that fatty acid metabolism, protein secretion, and xenobiotic metabolism scores were the highest in the “CT rec” group and the lowest in “ET rec” group. ESTROGEN_RESPONSE_EARLY score was highest in the “No rec” group. In analyzing the relationship amongst the three groups in breast TIME, the immune cytolytic activity scores were the highest in the “CT rec” group. High risk of an RPM tumor was indeed correlated with high immune cytolytic activity scores (Fig. 4). Regarding the immune cell composition, immunostimulatory cells, M1 macrophages were higher in the “CT rec” group compared with the “No rec” group, and follicular helper T cells were lower in the “CT rec” group compared with the “ET rec” group. Immunosuppressive Tregs were the lowest in the “CT rec” group. Additionally, monocytes were found to be higher in the “CT rec” group compared with the “ET rec” group. These results indicate that high-risk tumors among patients in the RPM were associated with poor treatment efficacy, which was supported by their correlation with pathway and TIME, which were involved in treatment resistance.
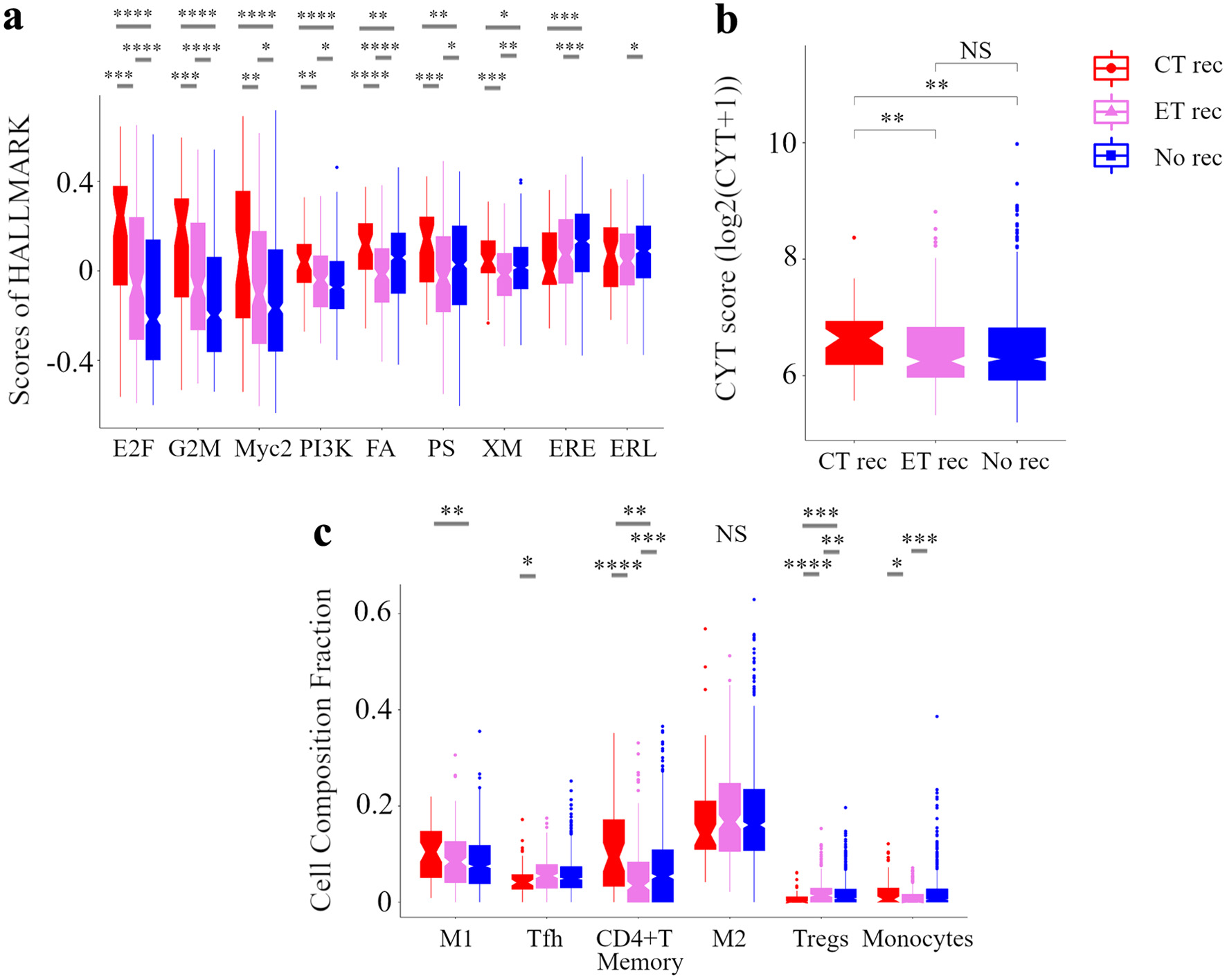 Click for large image | Figure 6. Analysis of the tumor microenvironment with no additional effect of chemotherapy and endocrine therapy. Box plots of the relationship between recurrence by treatments and signaling pathways in GSVA (a) and CYT (b) and immune cell composition (c) in METABRIC HR+ HER2- cohort are shown. Based on treatments and recurrence, we classified patients into following three categories: a group of patients who were treated with ET and CT but relapsed as CT rec, a group of patients who were treated with ET alone but relapsed as ET rec, and a group of patients who were treated with ET with or without CT and not relapsed as No rec. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. METABRIC: Molecular Taxonomy of Breast Cancer International Consortium; GSVA: Gene Set Variant Analysis; CYT: immune cytolytic activity; ET: endocrine therapy; CT: chemotherapy; E2F: E2F_TARGETS; G2M: G2M_CHECKPOINT; Myc2: MYC_TARGETS_v2; PI3K: PI3K_AKT_MTOR_SIGNALING; FA, FATTY_ACID_METABOLISM; PS: PROTEIN_SECRETION; XM: XENOBIOTIC_METABOLISM; ERE: ESTROGEN_RESPONSE_EARLY; ERL: ESTROGEN_RESPONSE_LATE; WNTβ: WNT_BETA_CATENIN_SIGNALING; M1: M1 macrophage; Tfh: follicular helper cells; M2: M2 macrophage; Tregs: CD4+ regulatory T cells. |
| Discussion | ▴Top |
Given the high prevalence and long latency of HR+ BC, the ability to predict prognosis is vital to selecting the optimal therapy for each patient and avoiding overtreatment [4]. Methods to better stratify individuals at high risk for BC development have been a focus of research interest for over a decade [2]. However, none of the tests developed to date are adequate predictors of survival. Therefore, we aimed to test the hypothesis that artificial intelligence can be used to develop a new framework for predicting prognosis in BC patients in multiple validation BC cohorts.
This study generated three interesting results with clinical implications. First, our RPM was able to stratify patients by prognosis in multiple cohorts. We first extracted 155 prognostic-related genes through a meta-analysis of all protein-coding genes associated with distant RFS in the METABRIC HR+ HER2- cohort (Supplementary Material 2, www.wjon.org). we combined the Cox-PH model with recursive feature elimination to narrow down the nine best predictors (Table 2). We next applied an LRM to develop tools that can accurately predict recurrence risk in BC patients based on the binary expression status of these nine genes.
Interestingly, most of these nine genes have not been well characterized to date with regard to their relation to basic or clinical oncology, with CYP4F22, TUBA3D, and HNMT apparently not having been studied in the field at all. Therefore, further research will be needed to elucidate the effects of nine genes based on RPM to extend the prognosis of BC patients. Finally, we found that our RPM can universally and predominantly stratify prognosis in all HR+ HER2- BC cohorts, using the Kaplan-Meier method and verified by the log-rank test (Fig. 2). In a similar study, Shimizu and Nakayama reported generating a prognostic score with a neural network for 23 genes narrowed down by one of the machine learnings, random forest, from about 20,000 genes [52]. The combination of the score and clinical stage suggested that prognosis could be stratified more precisely and that unnecessary CT could be avoided.
Further, the time-dependent AUC analysis of RPM showed a trend towards a significant decrease in accuracy around 10 years, although there was some variability between cohorts (Supplementary Material 4, www.wjon.org). In general, the HR+ BC subtype has a longer recurrence period than the HER2-enriched or triple-negative subtypes. Thus, prediction of late recurrence is one of the most important factors in predicting recurrence of HR+ BC [5, 16]. Therefore, to further improve the accuracy of our model, it is necessary to create a model that considers not only early recurrence, but late recurrence as well.
Second, the high-risk group in the RPM was associated with major BC therapeutic target pathways and TIME. We have shown that cell cycle-associated gene sets, MYC targets, and PI3K-AKT-mTOR signaling in GSEA were enriched in the high-risk group in the RPM (Fig. 3). We and many other research groups have previously reported that genetic mutations that cause abnormalities in the PI3K-AKT-mTOR pathway are closely related to the prognosis of BC patients [53-58]. Further, recent advances in sequencing of the human genome have revealed cell cycle and PI3K-AKT-mTOR pathway as therapeutic targets for HR+ HER2- metastatic BC, ranging from hormone therapy single agents to CDK4/6 inhibitors (palbociclib, ribociclib, abemaciclib), and an mTOR inhibitor (everolimus) [59, 60]. The cell cycle has also been identified as an important therapeutic target in primary BC as the monarchE trial demonstrated that abemaciclib was particularly useful as an adjuvant therapy [61].
In the component analysis of TME, the high-risk group in the RPM was associated with high levels of immune cells from the lymphoid and myeloid lineage and immune cytolytic activity, as well as low levels of stem and stromal cells (Fig. 4). Tumor-infiltrating lymphocytes are broadly classified into CD4+ helper cells, Tregs, and effector cells such as natural killer cells and CD8+ T cells [62]. Tregs, which normally suppress autoreactive T cells, suppress anti-tumor responses in TME and are therefore a poor prognostic factor in BC [63, 64]. Conversely, infiltration of CD8+ effector T cells into tumors is associated with longer BC-specific survival, independent of other prognostic factors such as tumor grade, clinical stage, and vascular invasion [63, 65]. Regarding the immune cytolytic activity score, which indicates the relative degree of anticancer immune activity, we have reported that it functions as a prognostic biomarker for BC [16, 23, 24]. Surprisingly, the high-risk group in the RPM had a mixture of good and poor prognostic factors, as shown by high correlations with immune cytolytic activity, Th1, Th2, Tregs, and the effector memory CD8+ T cells.
On the other hand, regarding myeloid-derived immune cells, macrophages are mostly of the M1 phenotype during normal immune response and are involved in the Th1 cytokine response to various pathogens. However, the tumor-associated macrophages that are formed in breast tumors and typically belong to the M2 phenotype, allow for cancer cell survival, and have been positioned as a poor prognostic factor in BC [62, 63]. Dendritic cells have different roles in tumors depending on their degree of maturation. Tumor-associated immature dendritic cells produce pro-angiogenic factors and actively promote tumor growth. Mature dendritic cells activate CD4+ and CD8+ T cells to attack tumor cells and reduce metastasis [62, 66]. Similar to lymphoids, the high-risk group in the RPM had a mixture of good and poor prognostic factors, as shown by high correlations with activated dendritic cells, M1 macrophages, and M2 macrophages. A possible reason for the discrepancy between the high-risk group in the RPM and immune cell function, is that immune response in tumor tissue is a series of carefully controlled events that can be optimally addressed as a group rather than as individual cells [67].
Third, the high-risk group in the RPM was associated with poor treatment efficacy, which was supported by their correlations with pathways and TIME involved in treatment resistance. We demonstrate that risk classification by RPM was associated with the effect of treatment, especially the effect of ET in HR+ HER2- BC patients (Fig. 5). In the analysis of the relationship between treatment outcome and signaling pathways, immune cytolytic activity, and immune cell composition, HR+ HER2- BC patients treated with CT but who relapsed had higher levels of E2F targets and G2M checkpoints, and were associated with lower levels of immunosuppressive M2 macrophages and Tregs (Fig. 6). According to a widely accepted idea, both cancer cell-specific properties, as well as signals derived from cells in TME play a critical role in cancer therapy response [68]. For example, Rosenfeldt and his colleagues showed that the E2F1-ABCG2 axis suppresses the CT-induced cell death that can be restored by the inhibition of ABCG2 [69]. Further, Velaei and colleagues reviewed the impact of conventional anticancer CT on the relationship between the tumor and the immune system as follows [68]. High levels of the tumor-infiltrating lymphocytes in the HR- HER2+ subtype are related with good response to CT. Above all, high levels of CD8+ cytotoxic T lymphocytes synergistically increase the effect of anthracycline or anthracycline/taxane based neoadjuvant CT and low levels of Tregs enhance CT response. In addition, targeting tumor-associated macrophages in combination with CT may improve the effect of CT, since macrophages induced by cytotoxic CT can protect tumor cells from death due to a cathepsin-dependent function. Focusing on the importance of gene expression profiles in the tumor stroma of BC patients, Finak et al generated a 26-gene prognostic predictor that predicts clinical outcomes regardless of clinical subtype [70]. Their model was correlated with two different sets of relevant genes: hypoxia and angiogenesis. These were associated with poor prognosis or displayed a Th1-like immune response associated with favorable outcomes. We demonstrate that high-risk status in the RPM was associated with poor treatment efficacy, which was supported by their correlation with E2F targets, G2M checkpoints, MYC targets, and PI3K-AKT-mTOR signaling, immune cytolytic activity, M1 macrophages, and Tregs, which were involved in treatment resistance. Further research is needed to create models that stratify prognosis by therapeutic factor in estrogen receptor (ER)+ HER2- BC and to identify novel strategies to overcome therapeutic resistance.
Although the study demonstrates promising results, it has limitations. First, this study utilized multiple large patient cohorts, and is therefore a retrospective study. Secondly, the investigated cohorts were not genetically analyzed using a common platform. Finally, the cohorts other than METABRIC lacked data on treatment regimens and the relationship between the RPM and treatment outcomes could not be investigated.
In conclusion, using machine learning, we identified nine genes related to BC prognosis from more than 18,000 genes and created an RPM. The RPM was able to stratify prognosis in multiple cohorts. This is because the RPM reflects major BC therapeutic target pathways and TIME and, further supported by the therapeutic effect of CT and ET in patients with BC. Based on these reported results, we anticipate that further studies can be conducted to better understand the mechanisms of recurrence and resistance to therapy in BC.
| Supplementary Material | ▴Top |
Suppl 1. Patients and clinical characteristics in TCGA HR+ HER2- cohort and other independent HR+ HER2- BC cohorts, GSE199135, GSE9195, GSE6532, and GSE21653. TCGA: The Cancer Genome Atlas; HR+: hormone receptor positive; HER2: human epidermal growth receptor 2; BC: breast cancer; PgR: progesterone receptor.
Suppl 2. 155 differentially expressed genes with |log2-fold change| > 0.25 and P < 0.05 in distant recurrence of HR+ HER2- breast cancer. HR+: hormone receptor positive; HER2: human epidermal growth receptor 2.
Suppl 3. 23 genes closely involved in distant RFS in HR+ HER2- patients extracted from 155 signature genes using the Cox-PH model. RFS: recurrence free survival; HR+: hormone receptor positive; HER2: human epidermal growth receptor 2; Cox-PH: Cox Proportional-Hazards.
Suppl 4. Analysis of changes in predictive accuracy of recurrence prediction model over clinical course. Time-dependent receiver operating characteristic curve depicting the time-dependent AUC values of the recurrence prediction model in METABRIC HR+ HER2- cohort, TCGA HR+ HER2- cohort, GSE199135, GSE9195, GSE6532, and GSE21653 are shown. AUC: area under curve; METABRIC: Molecular Taxonomy of Breast Cancer International Consortium; HR+: hormone receptor positive; HER2: human epidermal growth receptor 2; TCGA: The Cancer Genome Atlas.
Acknowledgments
None to declare.
Financial Disclosure
This work was supported by NIH grant R01CA160688 to KT, and NCI grant P30CA016056 involving the use of Roswell Park Cancer Institute’s Bioinformatics and Biostatistics Shared Resources.
Conflict of Interest
All of the authors declare that they have no actual, potential, or perceived conflict of interest regarding the manuscript submitted for review.
Informed Consent
Not applicable.
Author Contributions
Takashi Takeshita: conceptualization, methodology, doftware, writing - original draft preparation, visualization. Hirotaka Iwase: Writing - reviewing and editing. Rongrong Wu: software, validation. Danya Ziazadeh: writing - reviewing and editing. Li Yan: data curation, software, validation. Kazuaki Takabe: supervision, funding acquisition.
Data Availability
The data presented in this study are available upon request from the corresponding author.
Abbreviations
BC: breast cancer; HR+: hormone receptor positive; HER2: human epidermal growth factor receptor 2; ET: endocrine therapy; CT: chemotherapy; METABRIC: Molecular Taxonomy of Breast Cancer International Consortium; TCGA: The Cancer Genome Atlas; GEO: Gene Expression Omnibus; Cox-PH: Cox proportional-hazards; RFS: recurrence free survival; LRM: logistic regression model; NES: normalized enrichment score; FDR: false discovery rate; GSVA: Gene Set Variation Analysis; TME: tumor microenvironment; DEGs: differentially expressed genes; AUC: area under the receiver operating characteristic curve; PgR: progesterone receptor; GSEA: Gene Set Enrichment Analysis; mTOR: mammalian target of rapamycin; Tregs: regulatory T cells; TIME: tumor immune microenvironment
| References | ▴Top |
- Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7-33.
doi pubmed - Esteva FJ, Sahin AA, Cristofanilli M, Arun B, Hortobagyi GN. Molecular prognostic factors for breast cancer metastasis and survival. Semin Radiat Oncol. 2002;12(4):319-328.
doi pubmed - Hayes DF. Clinical utility of genetic signatures in selecting adjuvant treatment: Risk stratification for early vs. late recurrences. Breast. 2015;24(Suppl 2):S6-S10.
doi pubmed - Kwa M, Makris A, Esteva FJ. Clinical utility of gene-expression signatures in early stage breast cancer. Nat Rev Clin Oncol. 2017;14(10):595-610.
doi pubmed - Puppe J, Seifert T, Eichler C, Pilch H, Mallmann P, Malter W. Genomic signatures in luminal breast cancer. Breast Care (Basel). 2020;15(4):355-365.
doi pubmed pmc - Zhang W, Zhao Z, Wang K, Shen L, Shi X. The International Conference on Intelligent Biology and Medicine (ICIBM) 2020: Scalable techniques and algorithms for computational genomics. BMC Genomics. 2020;21(Suppl 11):831.
doi pubmed pmc - Munir A, Vedithi SC, Chaplin AK, Blundell TL. Genomics, computational biology and drug discovery for mycobacterial infections: fighting the emergence of resistance. Front Genet. 2020;11:965.
doi pubmed pmc - Becker M, Schultze H, Bresniker K, Singhal S, Ulas T, Schultze JL. A novel computational architecture for large-scale genomics. Nat Biotechnol. 2020;38(11):1239-1241.
doi pubmed - Krop I, Ismaila N, Andre F, Bast RC, Barlow W, Collyar DE, Hammond ME, et al. Use of biomarkers to guide decisions on adjuvant systemic therapy for women with early-stage invasive breast cancer: American Society of Clinical Oncology Clinical Practice Guideline Focused Update. J Clin Oncol. 2017;35(24):2838-2847.
doi pubmed pmc - Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486(7403):405-409.
doi pubmed pmc - Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163(2):506-519.
doi pubmed pmc - Gupta A, Mutebi M, Bardia A. Gene-expression-based predictors for breast cancer. Ann Surg Oncol. 2015;22(11):3418-3432.
doi pubmed - Kamel HFM, Al-Amodi H. Exploitation of gene expression and cancer biomarkers in paving the path to era of personalized medicine. Genomics Proteomics Bioinformatics. 2017;15(4):220-235.
doi pubmed pmc - Katsuta E, Yan L, Nagahashi M, Raza A, Sturgill JL, Lyon DE, Rashid OM, et al. Doxorubicin effect is enhanced by sphingosine-1-phosphate signaling antagonist in breast cancer. J Surg Res. 2017;219:202-213.
doi pubmed pmc - Young J, Kawaguchi T, Yan L, Qi Q, Liu S, Takabe K. Tamoxifen sensitivity-related microRNA-342 is a useful biomarker for breast cancer survival. Oncotarget. 2017;8(59):99978-99989.
doi pubmed pmc - Takeshita T, Yan L, Asaoka M, Rashid O, Takabe K. Late recurrence of breast cancer is associated with pro-cancerous immune microenvironment in the primary tumor. Sci Rep. 2019;9(1):16942.
doi pubmed pmc - Takeshita T, Asaoka M, Katsuta E, Photiadis SJ, Narayanan S, Yan L, Takabe K. High expression of polo-like kinase 1 is associated with TP53 inactivation, DNA repair deficiency, and worse prognosis in ER positive Her2 negative breast cancer. Am J Transl Res. 2019;11(10):6507-6521.
pubmed pmc - Asaoka M, Patnaik SK, Zhang F, Ishikawa T, Takabe K. Lymphovascular invasion in breast cancer is associated with gene expression signatures of cell proliferation but not lymphangiogenesis or immune response. Breast Cancer Res Treat. 2020;181(2):309-322.
doi pubmed pmc - Tokumaru Y, Oshi M, Katsuta E, Yan L, Satyananda V, Matsuhashi N, Futamura M, et al. KRAS signaling enriched triple negative breast cancer is associated with favorable tumor immune microenvironment and better survival. Am J Cancer Res. 2020;10(3):897-907.
pubmed pmc - Asaoka M, Ishikawa T, Takabe K, Patnaik SK. APOBEC3-Mediated RNA editing in breast cancer is associated with heightened immune activity and improved survival. Int J Mol Sci. 2019;20(22):5621.
doi pubmed pmc - Katsuta E, Maawy AA, Yan L, Takabe K. High expression of bone morphogenetic protein (BMP) 6 and BMP7 are associated with higher immune cell infiltration and better survival in estrogen receptor-positive breast cancer. Oncol Rep. 2019;42(4):1413-1421.
doi pubmed pmc - McDonald KA, Kawaguchi T, Qi Q, Peng X, Asaoka M, Young J, Opyrchal M, et al. Tumor heterogeneity correlates with less immune response and worse survival in breast cancer patients. Ann Surg Oncol. 2019;26(7):2191-2199.
doi pubmed pmc - Takeshita T, Torigoe T, Yan L, Huang JL, Yamashita H, Takabe K. The impact of immunofunctional phenotyping on the malfunction of the cancer immunity cycle in breast cancer. Cancers (Basel). 2020;13(1):110.
doi pubmed pmc - Takeshita T, Tokumaru Y, Oshi M, Wu R, Patel A, Tian W, Hatanaka Y, et al. Clinical relevance of estrogen reactivity in the breast cancer microenvironment. Front Oncol. 2022;12:865024.
doi pubmed pmc - Osipowski P, Pawelkowicz M, Wojcieszek M, Skarzynska A, Przybecki Z, Plader W. A high-quality cucumber genome assembly enhances computational comparative genomics. Mol Genet Genomics. 2020;295(1):177-193.
doi pubmed - Alberghina L, Piccialli G. From computational genomics to systems metabolomics for precision cancer medicine and drug discovery. Pharmacol Res. 2020;151:104479.
doi pubmed - Lacorte GA, Machado MA, Martinez ML, Campos AL, Maciel RP, Verneque RS, Teodoro RL, et al. DGAT1 K232A polymorphism in Brazilian cattle breeds. Genet Mol Res. 2006;5(3):475-482.
pubmed - Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. 2020;17(3):261-272.
doi pubmed pmc - Hayward P. Word recognition localised to left occipitotemporal cortex. Lancet Neurol. 2006;5(6):475.
doi pubmed - Renfree SP, Makovicka JL, Chung AS. Risk factors for delay in surgery for patients undergoing elective anterior cervical discectomy and fusion. J Spine Surg. 2019;5(4):475-482.
doi pubmed pmc - http://www.cbioportal.org/.
- https://www.ncbi.nlm.nih.gov/geo/.
- https://www.python.org.
- https://numpy.org.
- https://scipy.org.
- https://pandas.pydata.org.
- https://seaborn.pydata.org.
- https://pypi.org/project/matplotlib/.
- https://www.r-project.org.
- Oshi M, Takahashi H, Tokumaru Y, Yan L, Rashid OM, Nagahashi M, Matsuyama R, et al. The E2F pathway score as a predictive biomarker of response to neoadjuvant therapy in ER+/HER2- breast cancer. Cells. 2020;9(7):1643.
doi pubmed pmc - Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401-404.
doi pubmed pmc - Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1.
doi pubmed pmc - Rueda OM, Sammut SJ, Seoane JA, Chin SF, Caswell-Jin JL, Callari M, Batra R, et al. Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature. 2019;567(7748):399-404.
doi pubmed pmc - Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173(2):400-416.e411.
doi pubmed pmc - Reynolds AP, Richards G, de la Iglesia B, Rayward-Smith VJ. Clustering rules: a comparison of partitioning and hierarchical clustering algorithms. Journal of Mathematical Modelling and Algorithms. 2006;5:475-504.
- http://software.broadinstitute.org/gsea/index.jsp.
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417-425.
doi pubmed pmc - Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220.
doi pubmed pmc - Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1-2):48-61.
doi pubmed pmc - Balli D, Rech AJ, Stanger BZ, Vonderheide RH. Immune cytolytic activity stratifies molecular subsets of human pancreatic cancer. Clin Cancer Res. 2017;23(12):3129-3138.
doi pubmed - http://bioconductor.org/.
- Shimizu H, Nakayama KI. A 23 gene-based molecular prognostic score precisely predicts overall survival of breast cancer patients. EBioMedicine. 2019;46:150-159.
doi pubmed pmc - Hortobagyi GN, Chen D, Piccart M, Rugo HS, Burris HA, 3rd, Pritchard KI, Campone M, et al. Correlative analysis of genetic alterations and everolimus benefit in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: results from BOLERO-2. J Clin Oncol. 2016;34(5):419-426.
doi pubmed pmc - Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Tomiguchi M, Sueta A, Murakami K, Iwase H. Clinical significance of plasma cell-free DNA mutations in PIK3CA, AKT1, and ESR1 gene according to treatment lines in ER-positive breast cancer. Mol Cancer. 2018;17(1):67.
doi pubmed pmc - Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Tomiguchi M, Sueta A, Iwase H. ESR1 and PIK3CA mutational status in serum and plasma from metastatic breast cancer patients: A comparative study. Cancer Biomark. 2018;22(2):345-350.
doi pubmed - Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Tomiguchi M, Sueta A, Murakami K, Omoto Y, et al. Comparison of ESR1 mutations in tumor tissue and matched plasma samples from metastatic breast cancer patients. Transl Oncol. 2017;10(5):766-771.
doi pubmed pmc - Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Tomiguchi M, Sueta A, Murakami K, Omoto Y, et al. Analysis of ESR1 and PIK3CA mutations in plasma cell-free DNA from ER-positive breast cancer patients. Oncotarget. 2017;8(32):52142-52155.
doi pubmed pmc - Takeshita T, Yamamoto Y, Yamamoto-Ibusuki M, Inao T, Sueta A, Fujiwara S, Omoto Y, et al. Prognostic role of PIK3CA mutations of cell-free DNA in early-stage triple negative breast cancer. Cancer Sci. 2015;106(11):1582-1589.
doi pubmed pmc - Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860-921.
doi pubmed - McAndrew NP, Finn RS. Clinical review on the management of hormone receptor-positive metastatic breast cancer. JCO Oncol Pract. 2022;18(5):319-327.
doi pubmed - Johnston SRD, Harbeck N, Hegg R, Toi M, Martin M, Shao ZM, Zhang QY, et al. Abemaciclib combined with endocrine therapy for the adjuvant treatment of HR+, HER2-, node-positive, high-risk, early breast cancer (monarchE). J Clin Oncol. 2020;38(34):3987-3998.
doi pubmed pmc - Zhang AW, Campbell KR. Computational modelling in single-cell cancer genomics: methods and future directions. Phys Biol. 2020;17(6):061001.
doi pubmed - Grosjean N, Blaby-Haas CE. Leveraging computational genomics to understand the molecular basis of metal homeostasis. New Phytol. 2020;228(5):1472-1489.
doi pubmed - Caroli J, Dori M, Bicciato S. Computational methods for the integrative analysis of genomics and pharmacological data. Front Oncol. 2020;10:185.
doi pubmed pmc - Krishnan A, Kloehn J, Lunghi M, Chiappino-Pepe A, Waldman BS, Nicolas D, Varesio E, et al. Functional and computational genomics reveal unprecedented flexibility in stage-specific toxoplasma metabolism. Cell Host Microbe. 2020;27(2):290-306.e211.
doi pubmed - Zhang C, Mathe E, Ning X, Zhao Z, Wang K, Li L, Guo Y. The International Conference on Intelligent Biology and Medicine 2019 (ICIBM 2019): computational methods and applications in medical genomics. BMC Med Genomics. 2020;13(Suppl 5):47.
doi pubmed pmc - Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1-10.
doi pubmed - Velaei K, Samadi N, Barazvan B, Soleimani Rad J. Tumor microenvironment-mediated chemoresistance in breast cancer. Breast. 2016;30:92-100.
doi pubmed - Rosenfeldt MT, Bell LA, Long JS, O'Prey J, Nixon C, Roberts F, Dufes C, et al. E2F1 drives chemotherapeutic drug resistance via ABCG2. Oncogene. 2014;33(32):4164-4172.
doi pubmed - Osipowski P, Pawelkowicz M, Wojcieszek M, Skarzynska A, Przybecki Z, Plader W. Correction to: A high-quality cucumber genome assembly enhances computational comparative genomics. Mol Genet Genomics. 2020;295(2):535.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.