

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 14, Number 6, December 2023, pages 540-550

Vasopressin Analog [V4Q5]dDAVP Exerts Cooperative Anticancer Effects in Combination With Low-Dose 5-Fluorouracil on Aggressive Colorectal Cancer Models
Natasha T. Sobola, b, d, Luisina M. Solernoa, b, d, Candela Llavonaa, b, Daniel F. Alonsoa, b, c ![]() , Juan Garonaa, b, c, e
, Juan Garonaa, b, c, e ![]()
aCenter of Molecular and Translational Oncology (COMTra), Unit of Translational Oncology, Science and Technology Department, National University of Quilmes, Buenos Aires, Argentina
bCenter of Translational Medicine, Unit of Biomedical Cancer Research (IBioCAN), Laboratory N° 6, El Cruce “Nestor Kirchner” Hospital, Buenos Aires, Argentina
cNational Council of Scientific and Technical Research (CONICET), Buenos Aires, Argentina
dThese authors contributed equally to the study.
eCorresponding Author: Juan Garona, Center for Molecular and Translational Oncology, National University of Quilmes, Buenos Aires, Argentina
Manuscript submitted August 22, 2023, accepted October 9, 2023, published online November 18, 2023
Short title: Effects of [V4Q5]dDAVP Plus 5-FU on CRC Models
doi: https://doi.org/10.14740/wjon1715
| Abstract | ▴Top |
Background: Colorectal cancer (CRC) is a leading cause of cancer-associated mortality worldwide. Despite being an essential component of systemic chemotherapy for advanced CRC, 5-fluorouracil (5-FU) clinical use has severe limitations, such as high toxicity, low selectivity and drug resistance. [V4Q5]dDAVP (1-deamino-4-valine-5-glutamine-8-D-arginine vasopressin) is a peptide vasopressin analog and a selective agonist of the arginine vasopressin type 2 membrane receptor (AVPR2), expressed in microvascular and tumor tissue. This synthetic compound has well-proven antitumor and antimetastatic activity in different tumor types, including metastatic CRC. The objective of this work was to assess the potential combinational benefits in preclinical CRC models after [V4Q5]dDAVP addition to 5-FU.
Methods: Effects on cellular viability, cell cycle progression, apoptosis and molecular mechanisms associated to [V4Q5]dDAVP treatment in combination with 5-FU were evaluated in murine CT-26 and human COLO-205 cell lines. In vivo, impact of dual therapy was explored on CRC tumor growth and metastatic spread.
Results: In CRC cells, [V4Q5]dDAVP (1 µM) addition to sub-IC50 5-FU concentrations resulted in the enhancement of cytostatic effects induced by chemotherapy. Reduction of cell viability after combined treatment was associated with cell cycle arrest in the G0/G1 phase, induction of apoptosis and increased gene expression of the cyclin-dependent kinase inhibitor p21 (CDKN1A) and the tumor suppressor p53 (TP53) in malignant cells, as assessed by flow cytometry, terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate (dUTP) nick end labeling (TUNEL), and quantitative reverse transcription polymerase chain reaction (qRT-PCR), respectively. In vivo, intravenous administration of [V4Q5]dDAVP (0.3 µg/kg) in combination with safe low doses of 5-FU (50 or 80 mg/kg for CT-26 or COLO-205 tumor models, respectively) effectively abrogated CRC growth, reducing aggressiveness of primary lesions and increasing survival of tumor-bearing mice. In addition, concomitant administration of [V4Q5]dDAVP and 5-FU inhibited pulmonary metastasis formation by CT-26 cells in immunocompetent mice, especially reducing macrometastatic disease.
Conclusions: [V4Q5]dDAVP seems to enhance the efficacy of 5-FU-based chemotherapy in CRC by modulating tumor progression, as well as metastatic dissemination, suggesting its potential role as a safe and cost-effective co-adjuvant agent for the management of advanced CRC.
Keywords: Colorectal cancer; [V4Q5]dDAVP; 5-FU; Antimetastatic; AVPR2; Co-adjuvant therapy
| Introduction | ▴Top |
Colorectal cancer (CRC) is the third most commonly diagnosed malignancy and the second cause of cancer-associated mortality, with incidence expected to increase over the next two decades [1]. Nearly one-fifth of the patients suffer from metastatic CRC (mCRC) at diagnosis, of which liver and lungs are the most frequent affected organs. Moreover, 50% of patients with localized disease will develop metastases after curative treatment. While survival rates have improved globally over the past years for CRC in general, stage IV disease portends a very poor prognosis, with a 5-year overall survival of approximately 15% [2].
In patients with advanced stage disease, cytotoxic therapy options and response remain limited. In particular, antimetabolite chemotherapeutic agent 5-fluorouracil (5-FU) represents one of the main constituents of cytotoxic combination regimens used for CRC management. Unfortunately, the 5-FU regimen is markedly associated with high toxicity and severe side effects, including life-threatening myelosuppression, gastrointestinal complications, and neurotoxicity [3]. In this scenario, the wide availability of various doses and treatment schedules of this antimetabolite allows clinicians to select specific 5-FU-based regimens with the most acceptable patterns of toxicity aiming at a more personalized approach. Combining 5-FU with other cytotoxic drugs, such as oxaliplatin, capecitabine, irinotecan and others, can enhance chemotherapy outcomes. However, the efficacy of these agents is still limited as many colorectal tumors develop chemoresistance [4]. As a result, identifying novel, effective, and well-tolerated therapeutic alternatives for their potential application when combined with 5-FU during CRC treatment is of paramount importance in order to overcome adverse effects and improve patient outcome.
Due to their ease of rational design, low production costs, high target specificity, and good tolerability, peptide-based drugs, including synthetic hormone derivatives, have enormous potential as therapeutic tools for cancer management. The peptide compound [V4Q5]dDAVP (1-deamino-4-valine-5-glutamine-8-D-arginine vasopressin) is a second-generation synthetic analog of naturally occurring antidiuretic hormone, also known as arginine vasopressin (AVP) [5]. This novel agent is a result of a structure-based drug design aiming to increase its biological activity [6, 7]. [V4Q5]dDAVP preserves several key structural features of its parental compound, and first-generation AVP derivative, desmopressin (dDAVP), a widely used hemostatic compound that has been repurposed for the management of different types of cancer as a result of its angiostatic and antimetastatic activity, with promising results in clinical trials for breast cancer surgery (NCT01606072) and symptom control in CRC patients with rectal bleeding (NCT01623206) [8-10]. [V4Q5]dDAVP, which is also a cyclic nonapeptide, retains dDAVP extended half-life and target-specificity as a result of a deaminated cysteine in position 1 and substitution of L-arginine with its D-enantiomer in position 8. In addition, this novel analog added second-generation sequence modifications at the N-terminal of the peptide such as replacement of glutamine by valine in position 4, enhancing the strength of ligand-receptor binding [11], and asparagine by glutamine in position 5, preventing aminoacidic deamination [12]. Both parental drug dDAVP and novel analog [V4Q5]dDAVP activate the adenylate cyclase/cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) axis after selective stimulation of arginine vasopressin type 2 membrane receptor (AVPR2), a receptor subtype which is expressed in microvascular endothelium, normal colonic tissue and colorectal malignant cells [13-15]. Therapeutic profile of the novel compound was evaluated using a large variety of clinically relevant experimental cancer models including, but not limited to, CRC, pancreatic adenocarcinoma, small cell and non-small cell lung cancer, melanoma and different molecular subtypes of malignant breast tumors. As a result, [V4Q5]dDAVP consistingly showed enhanced cytostatic, antimetastatic and angiostatic effects in comparison to parental peptide dDAVP [5]. Particularly in CRC, in a previosuly published study, we showed that direct cytostatic action and clonogenic growth inhibition by [V4Q5]dDAVP was strictly AVPR2-mediated and that sustained intravenous (IV) administration of the peptide at clinically relevant doses was capable of reducing CRC-driven angiogenesis and metastatic dissemination and outgrowth in both liver and lung, without overt signs of toxicity [16].
As previously described, considering that cytotoxic efficacy in CRC is often limited by tumor drug resistance caused by prior exposure, and risk of cumulative toxicities due to lack of target selectivity, the potential combination with novel highly selective cytostatic agents such as [V4Q5]dDAVP is highly interesting. The main objective of the present work was to assess the antineoplastic activity of [V4Q5]dDAVP in addition to low-dose 5-FU on aggressive experimental CRC models, characterizing the impact of dual therapy on key biological events in this disease, such as cancer cell growth and survival, cell cycle progression, apoptosis induction, tumor progression and metastatic spread.
| Materials and Methods | ▴Top |
Cell culture conditions and reagents
KRASG12C-mutant and highly metastatic CT-26 mouse colon carcinoma cells and BRAFV600E-mutant COLO-205 human colon adenocarcinoma cell line, were obtained from the American Type Culture Collection (ATCC CRL-2638 and CCL-222, respectively), and were maintained in Roswell Park Memorial Institute 1640 culture medium (RPMI-1640, Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS), 2 mM glutamine and 80 µg/mL gentamicin, in a humidified incubator (37 °C, 5% CO2). All tumor cells were harvested using a 0.025% trypsin/0.01% EDTA solution (Gibco) diluted in phosphate-buffered saline (PBS). [V4Q5]dDAVP (Mpr-Tyr-Phe-Val-Gln-Cys-Pro-DArg-Gly-NH2) was purchased from the American Peptide Company Inc. (BACHEM Group; lot number 1507008T). Peptide purity was 99% as assessed by high-performance liquid chromatography-mass spectrometry. [V4Q5]dDAVP was diluted in PBS for in vitro and in vivo studies. The chemotherapeutic agent 5-FU was provided by Microsules S.A.
Tumor cell viability
For viability assays COLO-205 or CT-26 cells were seeded in 96-well plates with 2.5 or 3.0 × 103 cells per well, respectively. After 72 h of exposure to treatment, MTS reagent ([3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium]) (Promega) was added to each well and absorbance at 490 nm was measured after 3 h.
Cell cycle distribution by flow cytometry
Semi-confluent cultures were starved for 24 h in serum-free medium and then treated with [V4Q5]dDAVP (1 µM), 5-FU (5 µM) or its combination for other 24 h. COLO-205 cells were harvested with trypsin/ethylenediaminetetraacetic acid (EDTA) solution, permeabilized and fixed using 70% v/v methanol for cytometric analysis. Washed cells were stained with a propidium iodide (PI) solution (50 µg/mL, Thermo Scientific), incubated for 30 min at 37 °C and 1 × 104 events were acquired with a FACSCalibur (BD Life Sciences) flow cytometer. Compensated data were analyzed and the distribution of cells in three major phases of the cycle (G0/G1, S and G2/M) was calculated with FlowJo v7.2 Software (BD Life Sciences).
Apoptosis
Detection of DNA fragmentation was evaluated by the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate (dUTP) nick end labeling (TUNEL) assay with an in situ cell death detection kit (DeadEnd™ Fluorometric TUNEL System, Promega) according to the supplier’s recommendations. Briefly, COLO-205 cells were seeded on coverslips, treated for 48 h with [V4Q5]dDAVP (1 µM), 5-FU (5 µM) or its combination, and then fixed with 4% paraformaldehyde for 15 min at room temperature, washed with PBS and permeabilized with 0.2% Triton X-100. Free DNA 3'-OH ends were labeled with fluorescein-12-dUTP, using a recombinant terminal deoxynucleotidyl transferase (rTdT) in a humidified chamber at 37 °C for 1 h. Cells were stained with DAPI-containing mounting medium (Vectashield Vectorlabs) and viewed under a fluorescence microscope (Cytation 5, BioTek). The ratios of viable/apoptotic cells for each group were quantified in 10 high-power fields (HPFs) of × 200 magnification. The number of apoptotic cells evaluated by TUNEL was expressed as (number of TUNEL-positive cells/total cells) × 100 in each specific condition (± 95% confidence limits), and analyzed by the χ2 test.
Quantitative real-time polymerase chain reaction (PCR)
Total RNA was extracted using the Easy Pure RNA Purification Kit (TransGen Biotech) according to the manufacturer’s recommendations and was reverse-transcribed with the Super Script III-First Strand transcriptase (Invitrogen). Quantitative real-time PCR was performed using the SYBRGreen PCR Master Mix (ThermoFisher) and the QuantStudio 3 qPCR system (Applied Biosystems). Relative quantification was calculated by the 2-ΔΔCt method, normalized to the endogenous HPRT1 gene expression. Multiple comparisons between experimental groups were conducted using mixed-effects analysis with Sidak’s test. The following target specific primers were used (forward and reverse, respectively): HPRT1, 5'-AACGTCTTGCTCGAGATGTG-3' and 5'-GCTTTGATGTAATCCAGCAGG-3; CDKN1A, 5'-GGCCGGGTTGTCGCCCTTTT-3' and 5'-GGCCGGGTTGTCGCCCTTTT-3'; and p53, 5'-GGATGCCTTTGTGGAACTGTAC-3' and 5'-TTCACTTGTGGCCCAGATAGG-3'.
Animals
Immunocompetent BALB/cAnNLAE and athymic nude NLAE:NIH (S)-Fox1nu female mice were purchased from UNLP (National University of La Plata, Buenos Aires, Argentina), with an age of 8 - 12 weeks and a weight of approximately 20 g. Animals were housed at five mice per cage with food and water provided ad libitum. All animal protocols have been carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the US National Institutes of Health (NIH Publications No. 8023, revised 1978) and were approved by our institutional Animal Care Committee UNQ-CICUAL (Resolution CD CyT No. 075/14). Results from animal studies were reported following ARRIVE guidelines.
Pulmonary experimental metastases
A total of 2 × 105 CT-26 cells in SFB-free RPMI medium were injected into the lateral tail vein of immunocompetent BALB/cAnNLAE mice. IV [V4Q5]dDAVP (0.3 µg/kg), alone or plus 5-FU-based chemotherapy, was administered twice, 30 min prior to tumor cell injection and 24 h later. After 72 h of CRC cell inoculation, three suboptimal intraperitoneal (IP) doses of 5-FU (50 mg/kg) were injected weekly on days 4, 11, and 18 post-tumor cell challenge. On day 21 mice were euthanized, lungs were recovered, weighed and fixed in 4% formaldehyde.
Histological assessment of metastatic lung colonization
After pulmonary lesions were confirmed by histopathology, color bright field images of the entire hematoxylin and eosin (H&E) stained lung sections were generated using the Cytation 5 Cell Imaging Multi-Mode Reader (BioTek Instruments) at × 2.5 magnification. A series of images were collected using a 4 × 5 grid and the stitching was performed using the Image Montage function of the Gen5 Image software, setting a tile overlapping of 10%. The quantification of metastatic versus healthy lung tissue was performed on the stitched images using the “Color Threshold’’ tool from the ImageJ 1.5p Software (NIH, USA), setting the units in “µm” using the scale bar in each photograph as reference.
CRC tumor progression protocols
First, human COLO-205 xenografts were established by subcutaneous (SC) injection of 2.5 × 106 cells in 0.3 mL of RPMI 1640 and Matrigel (BD Biosciences) mixture (1:1 volume ratio), in the flanks of female NLAE:NIH (S)-Fox1nu mice. Tumor latency was monitored by periodic palpation and treatment started at day 9, when tumor engraftment was confirmed in 100% of the animals. Chemotherapeutic 5-FU (80 mg/kg) was administered IP once a week for 3 weeks, alone or in combination with [V4Q5]dDAVP (0.3 µg/kg, IV), given three times per week until animal sacrifice. Tumor diameters (length and width) were recorded three times per week using a digital caliper for tumor growth assessment. Xenograft volume was estimated using the formula π/6 × Length × Width2. During the tumor progression protocols, animal weights and tumor growth rates (TGRs) were also assessed. TGRs represent the slopes of the linear regressions of tumor growth curves over time. In addition, the effect of combinational therapy on survival of COLO-205 xenograft-bearing mice was also evaluated. Log-rank test and Cox regression analysis were applied to establish the association of each treatment with survival. Xenograft-bearing mice were euthanized by cervical dislocation when humane tumor burdens of 1,500 mm3 were reached. When CRC xenografts exhibited early signs of ulceration (day 29), incidence of skin infiltration was analyzed using a non-parametric one-tailed binomial proportions test (setting the expected outcome for skin infiltration at 50%) and animals were photographed at high resolution.
Secondly, to generate CRC tumors in immunocompetent hosts, 1 × 105 CT-26 cells were injected SC in syngeneic BALB/c mice. Treatment started 20 days later, after all incipient tumors were detected by palpation. The 5-FU was administered using weekly IP injections at a dose of 50 mg/kg, alone or in combination with [V4Q5]dDAVP (administered as mentioned previously), until the end of the protocol at day 36.
Statistical analysis
Statistical analysis was performed using the GraphPad Prism v8.0.0 (GraphPad Software Inc.) [17]. In order to compare differences between two experimental groups, Mann-Whitney or t-tests were used for non-parametric or normal distribution of data, respectively. In case of more than two experimental groups, analysis of variance (ANOVA) analysis with Tukey’s multiple comparisons post-test was used when normal distribution of data was determined. Kruskal-Wallis analysis with Dunn’s multiple comparisons post-test was used in case of non-parametric distribution of data. Data corresponds to at least two or three independent experiments unless stated otherwise. Data were presented as mean ± standard deviation (SD) or standard error of mean (SEM), unless stated otherwise. Differences were considered statistically significant at a level of P < 0.05.
| Results | ▴Top |
First, we addressed the characterization of the in vitro antineoplastic activity after [V4Q5]dDAVP addition to 5-FU-based chemotherapy and the molecular mechanisms associated to such combinational effects. Evaluation of potential benefits of [V4Q5]dDAVP plus suboptimal concentrations of cytotoxic therapy was conducted on exponentially growing human and murine CRC cells. As shown in Figure 1a, IC50 values of 5-FU on COLO-205 and CT-26 cells were found as 24.7 and 1.4 µM, respectively. [V4Q5]dDAVP at 1 µM as a single agent and specially in combination with 5-FU at IC25-35 (5 or 0.5 µM for COLO-205 and CT-26 cells) significantly impaired CRC cell viability, reducing metabolic activity in malignant cells by up to 56% (Fig. 1b) (P < 0.0001). These results were in accordance with previously reported data, in which [V4Q5]dDAVP plus taxane-, alkylating- or antimetabolite-based therapy deployed synergistic inhibitory activity of cellular growth on high density cultures of aggressive breast and colorectal carcinomas [16, 18].
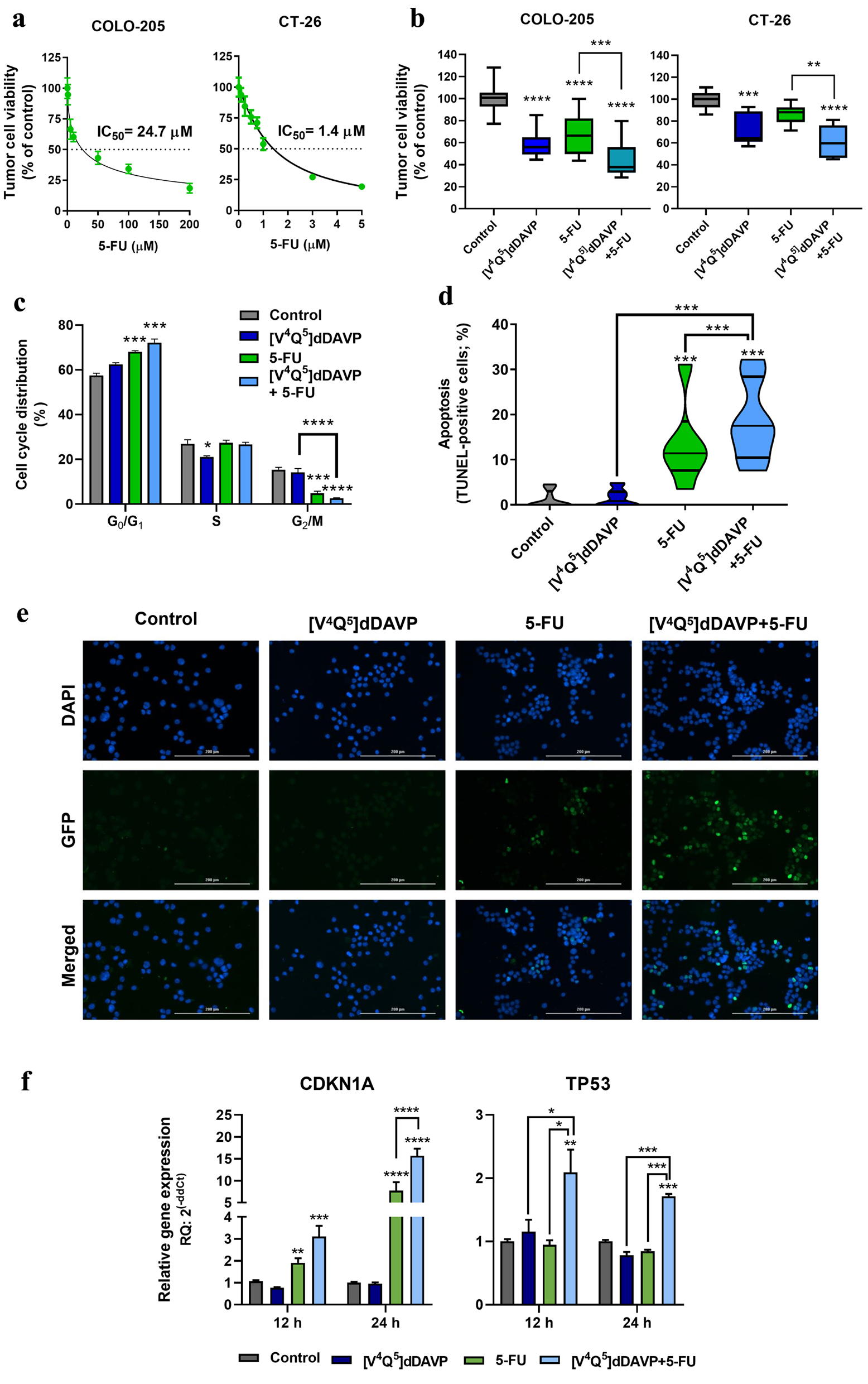 Click for large image | Figure 1. In vitro cytostatic activity of [V4Q5]dDAVP in combination with 5-fluorouracil on colorectal cancer cells and its impact on apoptosis induction and cell cycle progression. (a) Cytotoxic activity of 5-fluorouracil on COLO-205 (left) and CT-26 (right) colorectal cancer cells and IC50 calculation after a 72-h exposure to chemotherapeutic agent. (b) Effect on colorectal cancer cell viability of [V4Q5]dDAVP (1 µM) addition to 5 µM 5-fluorouracil for COLO-205 cells (left) or 0.5 µM for CT-26 cells (right). Direct cytotoxic/cytostatic effects on cancer cells were assessed by the metabolic MTS assay. (c) Cell cycle phase distribution evaluated by flow cytometry for COLO-205 cells after 24 h treatment with [V4Q5]dDAVP (1 µM), 5-fluorouracil (5 µM) or dual combined therapy. (d) Percentage of apoptotic cells after 48 h treatment with [V4Q5]dDAVP (1 µM), 5-fluorouracil (5 µM) or its combination in human colorectal cancer cell cultures. Apoptosis was assessed by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate (dUTP) nick end labeling (TUNEL). (e) Representative images of TUNEL labeling in human COLO-205 cell cultures under different treatments (× 200 magnification. Scale bar = 200 µm). (f) Relative gene expression of cyclin-dependent kinase inhibitor p21 (CDKN1A) and the tumor suppressor p53 (TP53) by quantitative reverse transcription polymerase chain (qRT-PCR) in colorectal cancer cells after 24 h treatment with [V4Q5]dDAVP and 5-fluorouracil, in combination or as monotherapies. Data are presented as mean ± standard error of mean (SEM) (a, c, f), box and whiskers with minimum to maximum values (b) or violin plots (d) and are representative of at least two or three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. |
The molecular mechanisms associated with the control of cell cycle progression and the induction of apoptosis are strongly dysregulated in CRC [19, 20]. Our group has previously reported that [V4Q5]dDAVP induces a partial arrest in the G0/G1 cell cycle phase on triple-negative breast cancer cells and that this effect is associated with the increase of intracellular cAMP and subsequent activation of PKA [6]. Furthermore, it has been previously reported that chemo-resistant CRC cells are addicted to low cAMP levels [21]. Following this rationale, we sought to investigate the impact of [V4Q5]dDAVP (1 µM) treatment plus 5-FU (5 µM) over the cell cycle progression of human CRC cells. After 24 h of drug incubation, combined treatment was associated with a significant cell cycle arrest in the G0/G1 phase, increasing the tumor cell population from 57.5% in the vehicle-treated group to 72.1% for [V4Q5]dDAVP + 5-FU (P < 0.001), as well as a sixfold decrease in the percentage of cells in the G2/M in comparison to control (Fig. 1c and Supplementary Material 1, www.wjon.org) (P < 0.0001). Interestingly, [V4Q5]dDAVP as a single was also capable of decreasing the percentage of cells in the synthesis phase of the cycle (S) (P < 0.05).
In addition, the ability of [V4Q5]dDAVP (1 µM), alone or in combination with chemotherapy, of inducing apoptosis in human CRC cells was also studied by fluorescence microscopy and the TUNEL assay. While after 48 h of treatment, TUNEL-positive rates for the control and [V4Q5]dDAVP groups were below 2%, combined treatment after addition of the novel synthetic analog to 5-FU (5 µM) caused a significant increase in the percentage of apoptotic cells from 1.2% to 18.45%, improving not only [V4Q5]dDAVP activity but also the pro-apoptotic effect of 5-FU as a monotherapy (13.45%) (Fig. 1d, e) (P < 0.001).
To further investigate the molecular mechanisms responsible for the ability of the drug combination to regulate tumor cell viability, cell cycle, apoptosis and survival, gene expression of the cyclin-dependent kinase inhibitor p21 (CDKN1A) and the tumor suppressor p53 (TP53) were studied using quantitative reverse transcription (RT)-PCR. After the first 12 h of drug exposure, a significant increase in p21 expression was observed as a consequence of 5-FU treatment (5 µM) (P < 0.01). After 12 h, [V4Q5]dDAVP (1 µM) addition resulted in a 15.7-fold gene expression increase versus control (P < 0.001), improving the effect of 5-FU as a single agent (7.7-fold increase in comparison to control group) (Fig. 1f, left) (P < 0.001). Moreover, the expression of p53 was rapidly stimulated and maintained as a result of the [V4Q5]dDAVP and 5-FU dual treatment, showing a 110% and 70% increase in the relative amount of transcripts found in the vehicle-treated CRC cells after 12 (P < 0.01) and 24 (P < 0.001) h of drug exposure, respectively.
Despite the fact that 5-FU continues to be the basis of adjuvant treatment for patients with mCRC, patient survival after treatment still remains poor and adverse effects are alarmingly common. In this setting, lung metastatic disease ranks as the second site of dissemination in CRC after liver, representing an important clinical challenge. For this reason, the effect of [V4Q5]dDAVP, alone or in combination with 5-FU, on CRC cell metastatic dissemination and lung colonization, was explored. For experimental metastasis studies, a suboptimal cytotoxic dose of 5-FU was selected for drug combinations with [V4Q5]dDAVP. As a result, [V4Q5]dDAVP (0.3 µg/kg IV) was administered 30 min before and 24 h after IV injection of mCRC cells, as a single agent or in addition to weekly doses of 5-FU (50 mg/kg IP) until the end of the protocol at day 21. As observed in Figure 2a, the therapeutic effects of the different compounds were first assessed on macrometastatic disease, after quantifying superficial pulmonary tumor nodules > 2 mm in diameter. Lungs belonging to saline-treated animals evidenced a high number of large metastatic lesions with a median of 11 macronodules per animal, with a range of 5 - 17 nodules. In contrast, concomitant administration of 5-FU with [V4Q5]dDAVP caused a 70% reduction in macrometastatic disease, versus control group (P < 0.001). Although not reaching statistical significance, suboptimal 5-FU treatment was associated to a partial inhibitory activity of 45% (P > 0.05). Finally, [V4Q5]dDAVP monotherapy resulted in a significant but smaller antimetastatic effect, reducing the median number of large metastatic lesions from 11 to 4, in comparison to animals treated with saline vehicle (P < 0.05). These results were confirmed after performing computer-assisted histopathological analysis of CRC cell-colonized lungs and measuring total metastatic burden (Fig. 2b), in which, once again, [V4Q5]dDAVP administration in combination with 5-FU resulted in greater therapeutic benefits (P < 0.0001). Representative images of H&E-stained sections of colonized lungs bearing subpleural and intrapulmonary mCRC nodules from different experimental groups are depicted in Figure 2c.
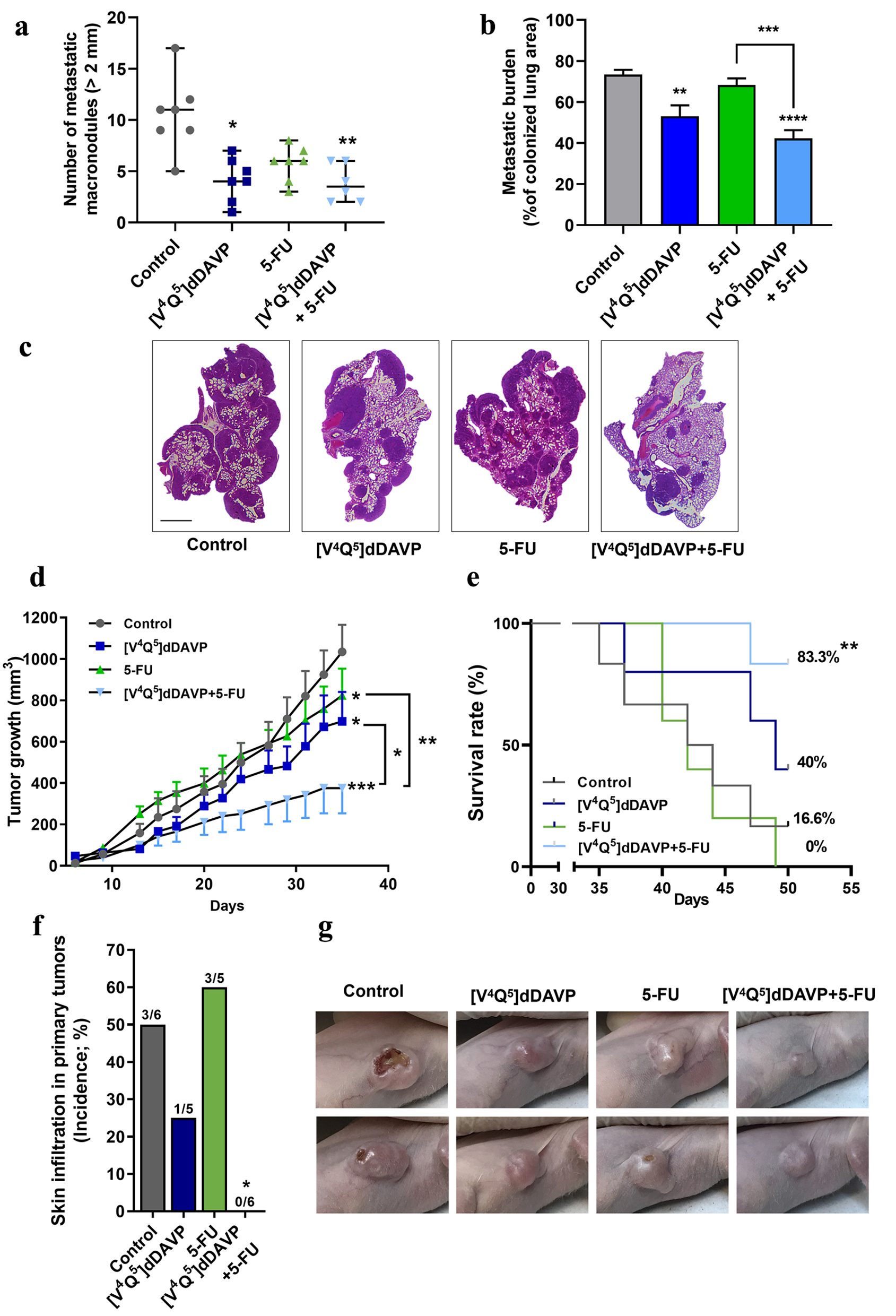 Click for large image | Figure 2. In vivo combinational effects of [V4Q5]dDAVP addition to low doses of 5-fluorouracil on colorectal cancer metastatic spread and tumor growth. Effects of combined therapy on CT-26 cell metastatic dissemination to lung (a, b and c) and COLO-205 xenograft progression (d, e, f and g) are shown. (a) Quantification of surface macrometastatic (> 1 mm of diameter) pulmonary lesions 21 days after highly metastatic CT-26 cells were injected intravenously (IV) in BALB/c mice. Animals were treated with [V4Q5]dDAVP (0.3 µg/kg IV) and 5-fluorouracil (50 mg/kg intraperitoneal (IP)), alone or in combination. (b) Metastatic burden in lungs was also assessed and quantified by computer-assisted histopathological analysis as area of CT-26 lung metastasis per total lung area. (c) Representative photographs of hematoxylin and eosin (H&E)-stained sections of lungs colonized by CT-26 metastatic cells from control mice receiving saline solution, [V4Q5]dDAVP or 5-fluorouracil alone, or [V4Q5]dDAVP plus 5-fluorouracil dual treatment. Scale bar = 2 µm. (d) Assessment of primary COLO-205 xenograft progression in nude mice. Curves representing tumor volume over time in mice receiving saline solution, [V4Q5]dDAVP (0.3 µg/kg IV), 5-fluorouracil (80 mg/kg IP) or its combination are shown. Statistical analysis was conducted on tumor growth rates calculated from day 10 to 31. (e) Effect of [V4Q5]dDAVP treatment, alone or co-administered with 5-fluorouracil-based chemotherapy, on survival of mice bearing COLO-205 xenografts. Kaplan-Meier survival plot for different experimental groups. (f) Incidence of skin infiltration in mice bearing COLO-205 xenografts treated with saline solution, [V4Q5]dDAVP, 5-fluorouracil (50 mg/kg IP) or dual concomitant therapy. (g) Representative high-resolution photographs of nude mice bearing COLO-205 primary tumors belonging to the different experimental groups were taken at day 29 of the colorectal cancer progression protocol and are representative of five or six animals per experimental group. Data are presented as scatter dot blots showing median with range (a), mean ± standard error of mean (SEM) (b, d) or percentages (e, f). *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. |
Having found both decreased growth and increased apoptosis in human CRC cells after [V4Q5]dDAVP addition to low-dose chemotherapy, we further explored the potential cooperative effects of the peptide analog plus 5-FU on tumor progression in vivo. COLO-205 xenografts were generated in athymic nude mice and treatments started after 9 days of tumor challenge and engraftment confirmation. The 5-FU was administered IP once a week for 21 days at a suboptimal 80 mg/kg IP dose, alone or in combination with [V4Q5]dDAVP at 0.3 µg/kg, IV, given three times per week. After 4 weeks of drug administration, despite all treatments were associated to an inhibition of CRC xenograft progression (mean TGR values were 37.1 ± 3.2, 25.9 ± 3.0, 26.4 ± 2.5, and 13.1 ± 2.8 mm3/day for control, [V4Q5]dDAVP (P < 0.05), 5-FU (P < 0.05) or [V4Q5]dDAVP + 5-FU (P < 0.001) groups, respectively), the combined treatment was capable of enhancing the anti-CRC activity of 5-FU (P < 0.05) and [V4Q5]dDAVP (P < 0.001) when administered as monotherapies, significantly reducing primary tumor burden by 57% against saline vehicle-treated group (1,034.6 ± 130.4 mm3 for control and 375.8 ± 122.1 mm3 for [V4Q5]dDAVP + 5-FU, mean ± SEM) (Fig. 2d). Cooperative inhibition of CRC growth was also assessed and confirmed in immunocompetent BALB/c mice bearing highly aggressive CT-26 syngeneic tumors, after administration of [V4Q5]dDAVP (also at 0.3 µg/kg, IV, three times per week) in addition to weekly IP doses of 5-FU (Supplementary Material 2, www.wjon.org) (P < 0.001).
The effect of [V4Q5]dDAVP treatment, alone or in combination with cytotoxic therapy, on survival of CRC xenograft-bearing animals was also evaluated. Despite low-dose 5-FU-based chemotherapy regimen ending after 4 weeks of treatment, [V4Q5]dDAVP administration continued until a tumor burden of 1,500 mm3 was reached and mice were euthanized. Sustained IV administration of [V4Q5]dDAVP in combination with 5-FU resulted in a significant increase in survival in comparison to control or single agent treatments (Fig. 2e) (P < 0.01). At day 50, survival fractions were 16.6%, 40%, 0% and 83% for control, [V4Q5]dDAVP, 5-FU and [V4Q5]dDAVP + 5-FU, respectively. Additionally, when the CRC xenografts exhibited signs of ulceration and necrosis, animals were photographed at high resolution and the incidence of skin infiltration was analyzed for each group (Fig. 2f). COLO-205 tumors corresponding to the vehicle-treated group showed invasive growth, causing visible ulceration and necrosis in 50% (3/3) of the experimental animals. Similarly, 60% (3/5) of the animals treated with 5-FU alone showed lesions in the superficial layer of the skin, but of a qualitatively smaller size, as shown in the representative images in Figure 2g. Animals treated with [V4Q5]dDAVP alone, or especially in combination with low-dose 5-FU, only showed a 20% incidence or a total inhibition of ulceration, respectively, indicating a strong modulation of local tumor aggressiveness (P < 0.05). Animal body weight variations, before and after treatments, and across different in vivo protocols were also assessed as a direct measure of treatment tolerability (Supplementary Material 3, www.wjon.org, for CT-26 experimental lung metastasis, COLO-205 xenograft progression and survival, and CT-26 tumor growth protocols, respectively). All evaluated therapies, including the [V4Q5]dDAVP + 5-FU combined treatment, were well tolerated, without significant changes in body weight or overt signs of toxicity (P > 0.05).
| Discussion | ▴Top |
To our knowledge, the present study establishes for the first time the efficacy and combinational effects of the synthetic peptide [V4Q5]dDAVP in addition to chemotherapeutic agent 5-FU using different CRC preclinical models. As previously described, despite the fact that 5-FU is a key antineoplastic drug in multiple chemotherapy combination regimens for CRC management, its use is widely associated with severe adverse effects and toxicity [3], as well as chemoresistance, even when administered in addition to other cytotoxic agents, such as oxaliplatin, irinotecan or capecitabine [4]. In this regard, exploring novel co-adjuvant drugs that have the capacity to selectively modulate different aspects of tumor aggressiveness and enhance the antitumor activity of chemotherapy, especially at lower and safer doses, is highly relevant.
CRC carcinogenesis and progression is widely associated with cell cycle deregulation and blockage of apoptosis induction, resulting in increased tumor cell survival and proliferative potential. In vitro we showed that [V4Q5]dDAVP addition to suboptimal 5-FU concentrations, results in cooperative cytostatic anti-CRC activity, reducing tumor cell viability and enhancing chemotherapy-induced apoptosis and cell cycle arrest. It is well described that [V4Q5]dDAVP direct cytostatic effects on tumor cells are AVPR2-dependent, as they were reverted by siRNA-mediated receptor knockdown or chemical blockade using specific receptor antagonists, and associated with AVPR2/AC/cAMP/PKA axis-related signaling pathways [6, 16]. cAMP/PKA pathway activation after exposure to 8-Br-cAMP analog or forskolin favors the arrest of tumor cell growth via extracellular signal-regulated kinase (ERK) inhibition and inhibits vascular mimicry and angiogenesis in CRC cells [22]. Interestingly, it has been previously shown that some chemo-resistant CRC cells are addicted to low cAMP levels, and that treatment with cAMP-elevating agents synergizes with phosphodiesterase (PDE)4/5 inhibitor rolipram, inducing mitogen-activated protein kinase (MAPK)-mediated apoptosis [21]. Here, it is also shown that short-term exposure to [V4Q5]dDAVP plus 5-FU treatment resulted in a significant increase in the expression of p53 and CDKN1A/p21. p53 modulates a large plethora of cellular responses such as DNA repair, cell survival, tumor metabolism, cell differentiation, among others, mainly acting as a central tumor suppressor [19]. CDKN1A/p21 is transcriptionally activated by p53, and is a main regulator of cell cycle progression, cellular senescence and stem cell aging [20]. In our study, p53 expression doubled in comparison to control or both monotherapies. When analyzing CDKN1A/p21, a dramatic 16-fold increase was observed in the dual therapy group, in contrast to vehicle-treated CRC cells, and a twofold increase against the chemotherapy-induced expression. In this setting, activation of AVPR2/AC/cAMP/PKA axis and associated downstream signaling pathways, may collaborate with and enhance the direct antineoplastic activity of antimetabolite 5-FU through different molecular mechanisms.
In the in vivo protocols, sustained treatment with [V4Q5]dDAVP in addition to low-dose 5-FU (50 or 80 mg/kg/day IP) significantly reduced the progression rates of CRC xenografts or syngenic tumors, enhancing therapeutic benefits and survival without apparent signs of toxicity. Increased antineoplastic activity as a result of [V4Q5]dDAVP addition to cytotoxic agents was previously reported for other tumor types, in which combination of this second-generation AVP analog to alkylating or taxane-based chemotherapy increased cytostatic effects of monotherapies, as well as decreased tumor growth and distant metastases in preclinical animal models [18]. Beyond the described molecular mechanisms of action in malignant cells associated with cell cycle arrest and apoptosis induction, efficacy of [V4Q5]dDAVP plus 5-FU dual therapy could also be associated with other indirect or stroma-associated events, such as angiogenesis inhibition. It is well known that high vascular density and overexpression of pro-angiogenic biomarkers in CRC correlate with disease progression [23]. The capactity of [V4Q5]dDAVP administration to modify in vivo angiogenesis triggered by human AVPR2-expressing CRC cells was previously evaluated in incipient tumor implants using basement membrane extract plugs in nude mice. Two weeks of IV treatment using [V4Q5]dDAVP was capable of reducing COLO-205-induced angiogenic response by nearly 60% in contrast with saline vehicle-treated animals [16]. Control of angiogenesis by [V4Q5]dDAVP seems to be associated with an increased cancer-mediated production of angiogenesis inhibitor ANG and a reduction of the expression of different proangiogenic markers such as platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF) and hypoxic-induced factor (HIF)-1α [9, 24]. On the other hand, several studies have shown that sustained administration of low and nontoxic doses of 5-FU or 5-FU prodrugs impairs angiogenesis in CRC tumors by favoring the production of thrombospondin-1 and decreasing VEGF expression [23]. Considering the angiostatic effects of [V4Q5]dDAVP, the combination of this AVPR2 novel agonist with sustained suboptimal treatment of 5-FU could favor a cooperative suppression of mitogenic potential of CRC cells as well as an inhibition of tumor-associated vasculature. Moreover, skin infiltration and superficial necrosis of human CRC xenografts were reduced in animals treated with [V4Q5]dDAVP, especially in addition to chemotherapy. These results are in line with previously published results by our group and others, in which exposure of malignant cells to first- and second-generation AVP analogs, alone [6, 9] or in combination with cytotoxic therapy [17, 25] resulted in a significant inhibition of local tumor aggressiveness. Reduction of migration and invasion after AVPR2 stimulation by [V4Q5]dDAVP, or parental compound desmopressin, were observed in a wide variety of tumor cell types, including breast, osteosarcoma, lung and prostate cancer. In this context, at the molecular level, modulation of urokinase plasminogen activator (uPA) and matrix metalloprotease (MMP)-2/MMP-9 secretion seems to be associated to such cooperative anti-invasive effects [6, 9, 18, 25].
With an about 44% and 10% prevalence, KRAS and BRAF mutations, respectively, are mostly studied in mCRC, as clinical reports demonstrated that first-line chemotherapy in combination with biologics targeting the epidermal growth factor receptor confers limited benefits. As a result, therapeutic response and prognosis of mCRC bearing KRAS or BRAF mutations are alarmingly poor [26]. Our work focused on different key biological aspects during tumor growth and metastatic progression, using highly aggressive and mCRC models with relevant molecular and mutational landscapes. For this study, we used the KRASG12C-mutant CT-26 mouse colon carcinoma cellular model and the human CRC COLO-205 cell line bearing the BRAFV600E-mutation for different in vitro and in vivo protocols. In particular, [V4Q5]dDAVP activity, given alone or as a dual therapy in addition to 5-FU, was tested on pulmonary metastatic spread and outgrowth by CT-26 cells. Besides being the second most common site of metastatic seeding in CRC, it has been reported that the lung metastatic niche is associated to decreased sensitivity to different cytotoxic agents, including 5-FU, being directly linked to multidrug resistance 1 (MDR1) overexpression by malignant disseminated cells [27]. It was previously shown that PKA activation, as observed after AVPR2 canonical activation, can modulate MDR1 activity [28]. Also, p53 and its transcriptional target p21, which are both upregulated after [V4Q5]dDAVP and 5-FU dual treatment, are known to reduce MDR1 expression, increasing drug sensitivity or even reverting drug resistance [29]. Moreover, hemostatic factor von Willebrand factor (vWF), a complex glycoprotein which is systemically released from AVPR2-expressing endothelial cells after [V4Q5]dDAVP administration, is known to modulate vascular inflammation and leucocyte recruitment, as well as angiogenesis and blood vessel normalization [30-32]. In this regard, other potential mechanisms associated to the release of vWF and other vascular mediators after [V4Q5]dDAVP administration, such as increased lymphocyte infiltration and immune function, or improved drug delivery into tumors, could be playing a role in the observed cooperative antimetastatic benefits and should be considered in the future. Integrating the above mentioned biological and molecular events, our hypothesis is that the antitumor activity observed after combining [V4Q5]dDAVP peptide to low doses of 5-FU is associated with both direct effects on AVPR2-expressing CRC cells as well as indirect stroma-mediated antineoplastic mechanisms, such as additive angiogenesis inhibition and vascular normalization, or even enhanced lymphocyte recruitment and effector function. In this scenario, cooperative cytostatic activity on tumor cells could be directly linked to AVPR2/cAMP/PKA axis activation by [V4Q5]dDAVP and enhanced expression of p53 and CDKN1A/p21, leading to cell cycle arrest, decreased survival and increased sensitivity to chemotherapy.
The present study confirmed an interesting anti-CRC activity of the evaluated peptide analog at IV doses of 0.3 µg/kg, alone or in addition to chemotherapy, showing no evident signs of toxicity, even in athymic animals administered up 18 times with tested compound. The observed exceptional tolerability is in line with previoulsy reported preclinical studies using different experimental animal models. In breast carcinoma-bearing mice, [V4Q5]dDAVP was administered intravenously 50 times using the same dose over a 3-month period of time with no clear signs of toxicity [18]. Moreover, in acute toxicology studies in naive Wistar rats, novel analog [V4Q5]dDAVP was well tolerated after being injected at 100 µg/kg doses, ≥ 300-fold above that required for antineoplastic activity [6]. However, despite its good safety profile, it should be born into mind that AVPR2 agonists such as [V4Q5]dDAVP or parental repurposed hemostatic agent dDAVP, also act on renal AVPR2 after systemic administration, modulating water reabsorption and hydrosaline balance. Considering that euvolemic hyponatremia was previously associated to tumor-secreted AVP in patients with syndrome of inappropriate antidiuretic hormone secretion [33] or after dDAVP administration in some cancer patients [8, 10], [V4Q5]dDAVP should be used under strict monitoring of fluid intake and output, as well as patient serum sodium levels.
Our work has several limitations worth noting. It is widely recognized that experimental animal models of CRC should recapitulate the tumor microenvironment as closer to the clinical setting as possible, considering its impact on disease progression and response to therapies [34]. It is known that AVP analogs such as [V4Q5]dDAVP target both cancer cells and different cellular components of its associated stroma. Taking that into account, the influence of CRC tumor microenvironment should not be neglected, and the use of orthotopic colorectal tumor models should be considered for future studies. In addition, it is known that CRC is characterized by high inter- and intra-tumor heterogeneity and complex genotypic landscapes. With the objective of reproducing this complex clinical heterogeneity, the use of CRC patient-derived xenografts in immunosuppressed animals should be addressed as a priority. Finally, considering its poor prognosis and elevated incidence, as well as relevant biological functions of the hepatic tissue, liver metastatic lesions associated to CRC attract particular interest of both clinicians and researchers [2]. Taking into account that [V4Q5]dDAVP was shown to be capable of impairing spread and growth of CRC cells in the liver [16], further studies should be conducted in the future assessing the impact of [V4Q5]dDAVP addition to standard-of-care cytotoxic agents on CRC liver metastasis. In this regard, the preclinical development of [V4Q5]dDAVP should also include combinational studies with oxaliplatin and irinotecan, in addition to 5-FU.
As a conclusion, this is the first study that demonstrated cooperative anti-CRC actions of the novel AVPR2-selective agonist [V4Q5]dDAVP in combination with non-toxic low doses of 5-FU. Considerable progress has been made in the treatment of CRC in recent years; however, challenges still remain, especially for advanced disease or aggressive and chemotherapy-refractory molecular subtypes. It is proposed that [V4Q5]dDAVP-based treatment strategy could be promising for the adjuvant setting of CRC management, potentially mitigating toxicities associated to high 5-FU doses and enhancing antineoplastic activities on both primary and metastatic lesions. Despite these encouraging results, additional research is still required to determine the precise therapeutic value in CRC management.
| Supplementary Material | ▴Top |
Suppl 1. Flow cytometry analysis and histograms showing distribution of tumor cells in the different phases of the cell cycle after 24 h in vitro exposure to [V4Q5]dDAVP (1 µM) added to suboptimal doses of 5-fluorouracil (5 µM).
Suppl 2. Assessment of primary CT-26 progression in BALB/c syngeneic mice. Curves representing tumor size over time in mice receiving saline solution, [V4Q5]dDAVP (0.3 µg/kg IV), 5-fluorouracil (50 mg/kg IP) or dual combined treatment are depicted. Statistical analysis was conducted on tumor growth rates calculated from day 20 to 36. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01 and ***P < 0.001.
Suppl 3. Body weight of animals belonging to CT-26 experimental lung metastasis (a), COLO-205 xenograft progression and survival (b), and CT-26 tumor growth (c) protocols. Weight variations at the end of the protocols are relativized to the mean body weight value registered prior to treatment initiation (taken as 100%). Data are presented as mean ± SEM.
Acknowledgments
None to declare.
Financial Disclosure
This work was supported by the National Agency for the Promotion of Science and Technology (ANPCYT, Argentina, grants No. PICT2017/2056 to DFA and JG, PICT2021/UCTH0005 to Dr. Alejandro Berra and JG, and PICT2021/AI00113 to Dr. Giselle Ripoll and JG), the National Institute of Cancer (INC, Argentina, grant No. INC2018/2021 to DFA and JG), the National Scientific and Technical Research Council (CONICET, Argentina, grant No. PIP11220210100708CO to DFA), and the National University of Quilmes (UNQ, Argentina, grant No. PUNQ1297/19).
Conflict of Interest
All authors have no conflict of interest to declare.
Informed Consent
Not applicable.
Author Contributions
NTS and LMS: formal analysis; investigation; methodology; validation. CL: investigation; methodology. DFA: conceptualization; funding acquisition; project administration; resources; software; writing - review and editing. JG: conceptualization; formal analysis; funding acquisition; investigation; project administration; supervision; validation; visualization; writing - original draft; writing - review and editing.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
| References | ▴Top |
- Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol. 2019;16(12):713-732.
doi pubmed - Cervantes A, Adam R, Rosello S, Arnold D, Normanno N, Taieb J, Seligmann J, et al. Metastatic colorectal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34(1):10-32.
doi pubmed - Ison G, Beaver JA, McGuinn WD, Jr., Palmby TR, Dinin J, Charlab R, Marathe A, et al. FDA approval: uridine triacetate for the treatment of patients following fluorouracil or capecitabine overdose or exhibiting early-onset severe toxicities following administration of these drugs. Clin Cancer Res. 2016;22(18):4545-4549.
doi pubmed - Blondy S, David V, Verdier M, Mathonnet M, Perraud A, Christou N. 5-Fluorouracil resistance mechanisms in colorectal cancer: From classical pathways to promising processes. Cancer Sci. 2020;111(9):3142-3154.
doi pubmed pmc - Garona J, Pifano M, Ripoll G, Alonso DF. Development and therapeutic potential of vasopressin synthetic analog [V(4)Q(5)]dDAVP as a novel anticancer agent. Vitam Horm. 2020;113:259-289.
doi pubmed - Garona J, Pifano M, Orlando UD, Pastrian MB, Iannucci NB, Ortega HH, Podesta EJ, et al. The novel desmopressin analogue [V4Q5]dDAVP inhibits angiogenesis, tumour growth and metastases in vasopressin type 2 receptor-expressing breast cancer models. Int J Oncol. 2015;46(6):2335-2345.
doi pubmed pmc - Pastrian MB, Guzman F, Garona J, Pifano M, Ripoll GV, Cascone O, Ciccia GN, et al. Structure-activity relationship of 1-desamino-8-D-arginine vasopressin as an antiproliferative agent on human vasopressin V2 receptor-expressing cancer cells. Mol Med Rep. 2014;9(6):2568-2572.
doi pubmed - Iseas S, Roca EL, O'Connor JM, Eleta M, Sanchez-Luceros A, Di Leo D, Tinelli M, et al. Administration of the vasopressin analog desmopressin for the management of bleeding in rectal cancer patients: results of a phase I/II trial. Invest New Drugs. 2020;38(5):1580-1587.
doi pubmed pmc - Ripoll GV, Garona J, Pifano M, Farina HG, Gomez DE, Alonso DF. Reduction of tumor angiogenesis induced by desmopressin in a breast cancer model. Breast Cancer Res Treat. 2013;142(1):9-18.
doi pubmed pmc - Weinberg RS, Grecco MO, Ferro GS, Seigelshifer DJ, Perroni NV, Terrier FJ, Sanchez-Luceros A, et al. A phase II dose-escalation trial of perioperative desmopressin (1-desamino-8-d-arginine vasopressin) in breast cancer patients. Springerplus. 2015;4:428.
doi pubmed pmc - Manning M, Misicka A, Olma A, Bankowski K, Stoev S, Chini B, Durroux T, et al. Oxytocin and vasopressin agonists and antagonists as research tools and potential therapeutics. J Neuroendocrinol. 2012;24(4):609-628.
doi pubmed pmc - Robinson AB, Rudd CJ. Deamidation of glutaminyl and asparaginyl residues in peptides and proteins. Curr Top Cell Regul. 1974;8(0):247-295.
doi pubmed - Kaufmann JE, Oksche A, Wollheim CB, Gunther G, Rosenthal W, Vischer UM. Vasopressin-induced von Willebrand factor secretion from endothelial cells involves V2 receptors and cAMP. J Clin Invest. 2000;106(1):107-116.
doi pubmed pmc - Monstein HJ, Truedsson M, Ryberg A, Ohlsson B. Vasopressin receptor mRNA expression in the human gastrointestinal tract. Eur Surg Res. 2008;40(1):34-40.
doi pubmed - Ripoll GV, Garona J, Hermo GA, Gomez DE, Alonso DF. Effects of the synthetic vasopressin analog desmopressin in a mouse model of colon cancer. Anticancer Res. 2010;30(12):5049-5054.
pubmed - Garona J, Sobol NT, Pifano M, Segatori VI, Gomez DE, Ripoll GV, Alonso DF. Preclinical efficacy of [V4 Q5 ]dDAVP, a second generation vasopressin analog, on metastatic spread and tumor-associated angiogenesis in colorectal cancer. Cancer Res Treat. 2019;51(2):438-450.
doi pubmed pmc - www.graphpad.com
- Garona J, Pifano M, Pastrian MB, Gomez DE, Ripoll GV, Alonso DF. Addition of vasopressin synthetic analogue [V(4)Q(5)]dDAVP to standard chemotherapy enhances tumour growth inhibition and impairs metastatic spread in aggressive breast tumour models. Clin Exp Metastasis. 2016;33(6):589-600.
doi pubmed - Oh HJ, Bae JM, Wen X, Jung S, Kim Y, Kim KJ, Cho NY, et al. p53 expression status is associated with cancer-specific survival in stage III and high-risk stage II colorectal cancer patients treated with oxaliplatin-based adjuvant chemotherapy. Br J Cancer. 2019;120(8):797-805.
doi pubmed pmc - Al-Maghrabi J, Al-Ahwal M, Buhmeida A, Syrjanen K, Sibyani A, Emam E, Ghanim A, et al. Expression of cell cycle regulators p21 and p27 as predictors of disease outcome in colorectal carcinoma. J Gastrointest Cancer. 2012;43(2):279-287.
doi pubmed - McEwan DG, Brunton VG, Baillie GS, Leslie NR, Houslay MD, Frame MC. Chemoresistant KM12C colon cancer cells are addicted to low cyclic AMP levels in a phosphodiesterase 4-regulated compartment via effects on phosphoinositide 3-kinase. Cancer Res. 2007;67(11):5248-5257.
doi pubmed - Wang S, Zhang Z, Qian W, Ji D, Wang Q, Ji B, Zhang Y, et al. Angiogenesis and vasculogenic mimicry are inhibited by 8-Br-cAMP through activation of the cAMP/PKA pathway in colorectal cancer. Onco Targets Ther. 2018;11:3765-3774.
doi pubmed pmc - Shi H, Jiang J, Ji J, Shi M, Cai Q, Chen X, Yu Y, et al. Anti-angiogenesis participates in antitumor effects of metronomic capecitabine on colon cancer. Cancer Lett. 2014;349(2):128-135.
doi pubmed - Ripoll G, Iannucci N, Giron S, Cascone O, Gomez D, Alonso D. Angiostatic activity of 1-Deamino-8-D-Arginine vasopressin and novel peptide analogues in breast cancer cells. AACR Meeting Abstracts. 2008;2008:295.
- Sasaki H, Klotz LH, Sugar LM, Kiss A, Venkateswaran V. A combination of desmopressin and docetaxel inhibit cell proliferation and invasion mediated by urokinase-type plasminogen activator (uPA) in human prostate cancer cells. Biochem Biophys Res Commun. 2015;464(3):848-854.
doi pubmed - Li ZN, Zhao L, Yu LF, Wei MJ. BRAF and KRAS mutations in metastatic colorectal cancer: future perspectives for personalized therapy. Gastroenterol Rep (Oxf). 2020;8(3):192-205.
doi pubmed pmc - Wilmanns C, Fan D, O'Brian CA, Bucana CD, Fidler IJ. Orthotopic and ectopic organ environments differentially influence the sensitivity of murine colon carcinoma cells to doxorubicin and 5-fluorouracil. Int J Cancer. 1992;52(1):98-104.
doi pubmed - Vanoye CG, Castro AF, Pourcher T, Reuss L, Altenberg GA. Phosphorylation of P-glycoprotein by PKA and PKC modulates swelling-activated Cl- currents. Am J Physiol. 1999;276(2):C370-378.
doi pubmed - Yang Y, Yuan H, Zhao L, Guo S, Hu S, Tian M, Nie Y, et al. Targeting the miR-34a/LRPPRC/MDR1 axis collapse the chemoresistance in P53 inactive colorectal cancer. Cell Death Differ. 2022;29(11):2177-2189.
doi pubmed pmc - Petri B, Broermann A, Li H, Khandoga AG, Zarbock A, Krombach F, Goerge T, et al. von Willebrand factor promotes leukocyte extravasation. Blood. 2010;116(22):4712-4719.
doi pubmed - Yin Q, Gu J, Qi Y, Lu Y, Yang L, Liu J, Liang X. ADAM28 from both endothelium and gastric cancer cleaves von Willebrand Factor to eliminate von Willebrand Factor-induced apoptosis of gastric cancer cells. Eur J Pharmacol. 2021;898:173994.
doi pubmed - Huszno J, Zembala-Nozynska E, Lange D, Kolosza Z, Nowara E. Answer to the Teresa Pusiol comments to the paper "The association of tumor lymphocyte infiltration with clinico-pathological factor and survival in breast cancer" by Huszno et al. Pol J Pathol. 2017;68(3):269.
doi pubmed - Onitilo AA, Kio E, Doi SA. Tumor-related hyponatremia. Clin Med Res. 2007;5(4):228-237.
doi pubmed pmc - Novoa Diaz MB, Martin MJ, Gentili C. Tumor microenvironment involvement in colorectal cancer progression via Wnt/beta-catenin pathway: Providing understanding of the complex mechanisms of chemoresistance. World J Gastroenterol. 2022;28(26):3027-3046.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.