

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Review
Volume 15, Number 1, February 2024, pages 28-37

Oxygen and Iron Availability Shapes Metabolic Adaptations of Cancer Cells
Rui Wanga, b, Aashiq Hussainc, Quan Quan Guoa, d, Xiao Wei Jina, f, Miao Miao Wange, f
aDepartment of Oncology, Suqian Affiliated Hospital of Xuzhou Medical University, Suqian City, China
bDepartment of Hematology, the Second Affiliated Hospital of Soochow University, Su Zhou City, China
cCancer Science Institute of Singapore, National University of Singapore, 119077 Singapore
dDepartment of Radiology, the Second Affiliated Hospital of Soochow University, Su Zhou City, China
eDepartment of General Surgery, Suqian Affiliated Hospital of Xuzhou Medical University, Suqian City, China
fCorresponding Author: Miao Miao Wang, Department of General Surgery, Suqian Affiliated Hospital of Xuzhou Medical University, Suqian City, China; Xiao Wei Jin, Department of Oncology, Suqian Affiliated Hospital of Xuzhou Medical University, Suqian City, China
Manuscript submitted October 1, 2023, accepted November 23, 2023, published online January 10, 2024
Short title: Iron-Deficiency Makes Cancer More Glycolytic
doi: https://doi.org/10.14740/wjon1739
- Abstract
- Introduction
- Oxygen Availability Induces Metabolic Adaptations
- Cooperation and Competition Between Glycolysis and OXPHOS
- Glycolysis Versus OXPHOS
- Some Tumor Cells Use Glutamine as an Energy Source
- Mutations of Tumor Suppressor Genes and Oncogenes Drive Cancer Towards Aerobic Glycolysis
- Clinical Relevance of Tumor Glycolysis and OXPHOS
- Iron Availability Shapes Electron Transfer and OXPHOS
- Oxygen Availability Regulates Lipid and Sugar Metabolic Pathways
- Closing Remarks and Future Directions
- References
| Abstract | ▴Top |
The dynamic changes between glycolysis and oxidative phosphorylation (OXPHOS) for adenosine triphosphate (ATP) output, along with glucose, glutamine, and fatty acid utilization, etc., lead to the maintenance and selection of growth advantageous to tumor cell subgroups in an environment of iron starvation and hypoxia. Iron plays an important role in the three major biochemical reactions in nature: photosynthesis, nitrogen fixation, and oxidative respiration, which all require the participation of iron-sulfur proteins, such as ferredoxin, cytochrome b, and the complex I, II, III in the electron transport chain, respectively. Abnormal iron-sulfur cluster synthesis process or hypoxia will directly affect the function of mitochondrial electron transfer and mitochondrial OXPHOS. More research results have indicated that iron metabolism, oxygen availability and hypoxia-inducible factor mutually regulate the shift between glycolysis and OXPHOS. In this article, we make a perspective review to provide novel opinions of the regulation of glycolysis and OXPHOS in tumor cells.
Keywords: Tumor; Glycolysis; OXPHOS; Iron; Metabolism; Oxygen
| Introduction | ▴Top |
Adenosine triphosphate (ATP) generated by mitochondrial oxidative phosphorylation (OXPHOS) provides sufficient energy necessary for normal cell metabolism. In contrast to normal cells, tumor cells exhibit an increase in glycolysis and a reduction in OXPHOS. Oncogenes, tumor suppressor genes, hypoxic microenvironment, mitochondrial DNA mutations, and other multifactorial effects were suggested to contribute to the tendency of cancer cells to use glycolysis. Normal cellular energy metabolism is characterized by the economical and efficient utilization of glucose in mitochondria for OXPHOS, whereas the energy metabolism of tumor cells is characterized by the active intake of glucose and aerobic glycolysis. This seemingly uneconomical energy supply method is necessary for tumor cells. Glycolysis provides energy and raw materials for biosynthesis and the continuous growth of tumor cells. This energy metabolic feature of tumor cells, also known as the Warburg effect, was observed by German scientist Otto Warburg as early as the 1920s [1].
Even within the same tumor, cancer is a heterogeneous disease and each tumor cell employs distinct metabolic processes. Although aerobic glycolysis is the primary metabolic pathway for tumor cells, mitochondrial OXPHOS still contributes a fraction of total cell ATP, and OXPHOS may play a crucial role in the development of certain types of tumors. The present review article provides an overview of the modulation of glycolysis and OXPHOS in tumor cells.
| Oxygen Availability Induces Metabolic Adaptations | ▴Top |
Before free oxygen appeared in the atmosphere on earth, eukaryotic cells mainly depended on glycolysis for energy. After oxygen appears in the atmosphere, glycolysis takes a back seat, and OXPHOS becomes the main way for cells to obtain energy because it is more effective than glycolysis and can produce more ATP; however, glycolysis, an ancient way of producing energy, has been preserved in the evolutionary process. Glycolysis plays an important role in the energy metabolism of organs or tissues such as the brain, liver, and muscles [2]. Glycolysis, which is the main energy supply way for anaerobic bacteria, is closely coupled with OXPHOS.
The glycolysis process takes place in the cytoplasm and only produces two ATP molecules, and subsequently, its final product, pyruvate, enters the mitochondria under aerobic conditions and is oxidized to acetyl-CoA (Ac-CoA), which condenses with oxaloacetic acid to form citric acid, starting the tricarboxylic acid (TCA) cycle and OXPHOS, which produces 36 ATP molecules [3]. Therefore, pyruvate acid is the raw material for the TCA cycle and OXPHOS. Under hypoxic conditions, pyruvate is reduced to lactic acid by lactate dehydrogenase A (LDH-A) and is exported outside the cell. Energy production is a response of cells to energy needs; therefore, the production of cellular ATP varies with the surrounding environment. Cells rely on glycolysis and OXPHOS to produce ATP, and for the ratio of these two energy production pathways depends on the environment and the cell. Under aerobic conditions, normal cells mainly rely on OXPHOS to produce ATP. This process provides 70% of the energy required for cell metabolism. Glycolytic activity is only enhanced under hypoxic conditions to compensate for the compromised OXPHOS function. Therefore, glycolysis and OXPHOS work together to maintain cell energy balance. Assuming that the total output for a cell is constant. If OXPHOS functions normally in tumor cells, they would not likely to rely on glycolysis for energy, i.e. normal OXPHOS could inhibit the activity of glycolysis so as to maintain cell energy balance. Conversely, if OXPHOS functions normally, it regulates the activity of glycolysis through different pathways, so as to maintain cell energy balance [4]. Different from normal cell metabolism, most tumors use glycolysis as the main way of energy production, both under aerobic and hypoxic conditions, which is called the Warburg effect.
Tumor cells differ from normal ones in their metabolism and energy supply. They can increase the uptake of glucose and glutamine and carry out aerobic glycolysis, resulting in more production of lactic acid and less ATP. Tumors are heterogeneous, in which each tumor has its own metabolic characteristics. Of note, the metabolism of tumor cells is not static, but constantly changes according to the microenvironment. These dynamic changes enable tumor cells to maintain selective growth advantages under unfavorable living conditions.
| Cooperation and Competition Between Glycolysis and OXPHOS | ▴Top |
Tumor cells vary in their origin and differentiation; hence, not all tumor cells show the glycolysis as dominant way of energy supply. Glycolysis contributes 2% to 64% of total ATP for tumor cells [5]. Suganuma et al [6] used 2-deoxy-D-glucose (2-DG), a glycolysis inhibitor, and oligomycin, an OXPHOS inhibitor to study the metabolism of four types of leukemia cells. NB4 cells are more sensitive to 2-DG than the other three cell lines; therefore, NB4 cells are considered to be glycolytic leukemia cells. The other leukemia cell THP-1 showed resistance to 2-DG but was sensitive to oligomycin; therefore, THP-1 cells are OXPHOS-dependent leukemia cells. These findings suggest that different tumors have different energy metabolism dominance.
Taken together, when using anti-metabolites to treat tumors, the metabolic characteristics of tumor cells need to be considered to maximize the efficacy of anti-metabolites.
Warburg believes that the reason for tumor cells to take aerobic glycolysis as the main energy metabolism method is that there is irreversible damage within tumor cell mitochondria, but currently there are different opinions on this point.
Studies have shown that there is no irreversible damage to the mitochondria of tumor cells. Glycolysis can inhibit OXPHOS. By inhibiting the glycolysis of tumor cells, mitochondrial OXPHOS can be restored [7-11]. For example, Fantin et al [7] observed that when the LDH-A is inhibited, OXPHOS activity can be enhanced, which can compensate for the reduced ATP due to glycolysis. This observation suggests that the active glycolysis of tumor cells is not due to mitochondrial OXPHOS defects, but because active glycolysis inhibits mitochondrial OXPHOS. Fantin et al also observed that when LDH-A was inhibited, the growth of tumor cells and tumorigenicity of mice decreased, indicating that the enhanced OXPHOS activity still cannot meet the needs of tumor cell growth, and hence LDH-A is a tumor drug target.
| Glycolysis Versus OXPHOS | ▴Top |
Glycolysis contributes to about 50-60% of the total ATP production for tumor cells [5]; hence, OXPHOS contributes a considerable proportion of total ATP output. Studies have shown that although human tumor cells (HL60, HeLa, 143B and U937) use mitochondrial OXPHOS to support their cell growth [12], this situation may change under hypoxic conditions. For the cervical HeLa cells and breast MCF-7 cells, the contribution of OXPHOS to cellular ATP is as much as 79% and 91% under normal circumstances, but under hypoxia, this contribution drops to 29% and 36%, respectively [13], suggesting that glycolysis tendency is more likely a result of hypoxia in these cell lines. Moreno-Sanchez et al [8] also pointed out in their retrospective review that although glycolysis is involved in tumor energy and plays an important role in total ATP output, a considerable variety of tumor cells still adopt OXPHOS as their main energy supply method, or a combination of glycolysis and OXPHOS. Under some conditions, the OXPHOS function of tumor cells even exceeds that of normal cells [14]. Recently, Singaporean researchers isolated the complete mitochondria from human ovarian cancer and peritoneal cancer cells. The succinate dehydrogenase, malate dehydrogenase and glutamine dehydrogenase activity can carry out OXPHOS and produce ATP, but relatively lower than normal cells [15].
According to metabolic features, Smolkova et al [11] divided the tumorigenesis into four stages. The first stage is stem cell transformation, which is mainly manifested by oncogene-mediated signal activation. The second stage is the hypoxia stage, in which signal pathways such as hypoxia-inducible factor (HIF), AMP-activated protein kinase (AMPK) and nuclear factor-kappa B (NF-κB) are activated. The results of the first and second stages of gene reprogramming resulted in Warburg effect, and OXPHOS was inhibited. The third stage is the low-glycemic stage. In this stage, the tumor grows so fast, and resulted in nutritional deficiency and activated the LKB1-AMPK-p53 pathway and (or) PI3K-Akt-mTOR pathway. The gene reprogramming leads to partial recovery of mitochondrial OXPHOS capacity, which is also related to the entry of Myc-mediated glutamine into the TCA cycle. The fourth stage is the recovery of mitochondrial OXPHOS function. This hypothesis suggests that the energy metabolism of cells is constantly changing in the process of transformation, and the reduction of mitochondrial function cannot discriminate cancer cells from normal counterparts.
The reverse Warburg supports tumor growth with OXPHOS function [14]. The reverse Warburg effect refers to the energy metabolism of cancer-associated fibroblasts (CAFs) around the tumor that turns to aerobic glycolysis under the action of cancer cells. The CAFs released lactic acid, ketone bodies or pyruvate can be used as an energy source by epithelial cancer cells and enter the TCA cycle for OXPHOS and ATP production [14-16]. In this case, tumors have formed an inseparable community with surrounding mesenchymal cells. This possibility exists because there is a co-evolution process between cancer cells and the interstitium. For example, under the “education” of tumor cells, the fibroblasts are transformed into CAFs, macrophages turned into tumor-associated macrophages (TAMs), neutrophils turned into tumor-associated neutrophils (TANs), and so on. These tumor-related mesenchymal cells are different from the original normal cells, in which there are genetic and epigenetic changes and their metabolism changed accordingly.
Recently, studies have pointed out that the lactic acid released by tumor cells due to hypoxia and enhanced glycolysis is not discharged as waste, but be used as energy raw materials for other tumor cells and surrounding mesenchymal cells. Also, LDH-B is converted into pyruvic acid under catalysis, which then enters the mitochondria and undergoes OXPHOS [14, 17]. Therefore, tumor cells communicate with surrounding cells through not only cytokines but also energy metabolites, in which they form a metabolic symbiosis of cells (metabolic symbiotic). These studies suggest that caution should be made when using lactic acid preparations for tumor patients, because of its possibility to promote tumor growth. These results provide a theoretical basis for using mesenchymal cells as tumor treatment targets.
While cancer cells retain the OXPHOS function, why do they tend to use aerobic glycolysis as their main metabolic pathway?
The main reasons are as follows: 1) Glycolysis provides the basis for the fast growth of tumor cells. Tumors grow faster than normal tissues, in which not only energy, but also biomacromolecules are needed for tumor growth. The intermediate products of glycolysis or truncated TCA cycle can be used by tumor cells for nucleotide acid synthesis, lipid, and protein synthesis [18]. 2) Although glycolysis produces less ATP compared with OXPHOS, it can produce ATP faster than OXPHOS, which is very suitable for the needs of fast-growing tumor cells [19]. Generally speaking, glycolysis can rapidly provide the energy needed by the rapid growth of tumor cells, and differentiated cells rely on OXPHOS [20]. For example, the glycolysis inhibitor 3-bromopyruvate (3-BP) is used to treat tumors. It is effective for fast-growing tumors but has little effect on slow-growing tumors. 3) Hypoxia and iron starvation is a common phenomenon in tumor tissues. Glycolysis just guarantees the selective growth advantage of tumor cells in this unfavorable microenvironment. The lactic acid produced by glycolysis is excreted outside the cell, acidifying the tumor microenvironment. This acidified microenvironment is not so good for normal tissues, but it provides advantages for tumor tissues. It can stimulate tumor cell growth, infiltration, and metastasis. 4) The reduced electron transport in the mitochondrial respiratory chain resulted in less production of reactive oxygen species (ROS) accordingly, which may be toxic to tumor cells.
Of course, tumor cells may retain OXPHOS function, which does not mean this mitochondrial OXPHOS is intact. The glycolysis of some tumors is indeed caused by damage to mitochondrial function [21, 22].
These damages include defects in the entry of pyruvate into mitochondria, truncated TCA cycles, reduced mitochondrial numbers and defects in the respiratory chain, increased inhibitors of ATP syntheses, increased sensitivity of mitochondrial DNA (mtDNA) to oxygen stress, etc. [8].
| Some Tumor Cells Use Glutamine as an Energy Source | ▴Top |
Although glucose is the main energy source for most tumors, it is not the only source of cellular energy. Researches have suggested that glutamine metabolism (glutaminolysis) may be another energy alternative pathway for some tumors [23, 24], because tumor cells can consume large amounts of glutamine [25]. In 1979, Reitzer et al [24] discovered that HeLa cells cultured in vitro used glutamine instead of sugar as an energy source , and other reports have indicated that glutamine can be used as an energy source by tumor cells [26, 27]. Unlike glucose, only certain tumor cells use glutamine as an energy source, not all tumor cells [28].
After glutamine enters the cell via the membrane transporter ASCT2, it is hydrolyzed into glutamate and ammonia under the action of glutaminase (GLS). Glutamate can interact with cysteine and glycine and form glutathione (GSH). GSH exists in almost all cells of the human body, participates in the redox regulation of human cells and is the main anti-oxidative enzyme in the cell [29]. Glutamine can be converted into α-ketoglutarate (α-KG), which then enters the TCA cycle for OXPHOS, and provides intermediate metabolites and energy to the cell. This situation is particularly obvious in the truncated TCA cycle, in which raw materials are injected into the TCA cycle; hence glutamine can provide energy for tumor cells when glucose cannot be used effectively [30]. The abovementioned observations demonstrated that tumor cells can use OXPHOS to produce ATP.
Notably, the increased use of glutamine in tumor cells is related to the activation of the oncogene Myc [31, 32]. Transfection of mouse embryonic fibroblasts (MEFs) with Myc can lead to a significant increase in the use of glutamine by MEFs. By contrast, when RNA interference is used to down-regulate Myc gene expression, the dependence of cells on glutamine is also reduced [32]. Other studies also support a metabolic relationship between glutamine and Myc. Human fibroblasts cultured with glucose-removed medium lead to cell death, and this process has nothing to do with Myc activity and is different from the mechanism of apoptosis; however, culturing human fibroblasts with a medium without glutamine can lead to Myc activity-dependent apoptosis [33], suggesting that for some tumors, blockade of glutamine metabolism may achieve better therapeutic effects than blockade of glucose. Notably, the situation in the body is much more complex. Cells can also use fatty acids and other substances for energy. The mutual substitution of glucose and glutamine is also seen in tumor cells. For example, when glucose supply is restricted, certain glycolytic glioma cells can switch from glycolysis to OXPHOS [34].
| Mutations of Tumor Suppressor Genes and Oncogenes Drive Cancer Towards Aerobic Glycolysis | ▴Top |
The reason why tumor cells undergo aerobic glycolysis, which is considered an inefficient method of energy production, may be closely related to oncogenes activation and tumor suppressor genes inactivation during tumorigenesis [35-37]. The mutation or overexpression of oncogenes Ras and Myc in malignant tumors is very common, and their activation drives the metabolic phenotype of tumor cells towards glycolysis. Ras can activate mTOR through the PI3K-Akt-mTOR signaling pathway [38], and mTOR can mediate the expression of HIF [39] to promote glycolysis. HIF is induced to adapt to the hypoxic microenvironment [40], and can up-regulate the enzyme activity of glycolysis [27]. In addition to mediating the expression of HIF [29], mTOR can also directly up-regulate glycolysis, from the uptake of glucose to the basic steps of glycolysis [40].
Excessive uptake of glucose exceeds the needs of tumor cells, and the result is that pyruvate is reduced to lactic acid and discharged outside the cell. Myc mainly affects the Warburg effect of tumor cells in two aspects. On the one hand, it promotes the activity of glycolysis-related genes, and on the other hand, it increases glutamine metabolism [41]. Myc is a transcription factor with a wide range of effects, which can affect up to 15% of human gene expression, including glycolysis and glucose transporter (GLUT) gene expression, such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH), hexokinase 1 (HK1), M2-type pyruvate kinase (PKM2), lactate dehydrogenase deficiency (LDH-A) and direct the energy metabolism of tumor cells towards the Warburg effect [41]. Myc can also promote the uptake and metabolism of glutamine in cancer cells, thus α-KG, a metabolite of glutamine, enters the TCA cycle for OXPHOS and provides energy for cells [42]. When Myc increases glutamine metabolism to provide energy, tumors reduce the use of glucose. Unlike Ras, which regulates glycolysis through the PI3K-Akt-mTOR signaling pathway, Myc does not rely on the PI3K-Akt signaling pathway to increase glutamine metabolism. Notably, PI3K-Akt signaling pathway inhibitors can reduce the utilization of glucose, but do not affect the utilization of glutamine [42]. Furthermore, when Ras and Myc affect tumor cell metabolism, a number of intermediate products of glycolysis or the TCA cycle also provide a large amount of raw materials for tumor cell biosynthesis [42, 43], which is also very important for the rapid growth of tumors.
Tumor suppressor protein tumor suppressor 53 (TP53) is a transcription factor with a wide range of biological functions, including cell energy metabolism. Notably, TP53 serves an important role in the balance between OXPHOS and glycolysis. Tumor cells with inactivated TP53 often show an increased glycolysis ratio. For example, in wild-type TP53 colon cancer HCT116 cells, the contribution of glycolysis to ATP is approximately 40%, whereas in TP53-mutant cells, the contribution of glycolysis to ATP is increased to approximately 66% [44]. There are numerous reasons for the increased proportion of glycolysis in response to TP53 inactivation. Under normal circumstances, TP53 downregulates the expression of GLUT genes GLUT1 and GLUT4, induces synthesis of cytochrome c oxidase 2 (SCO2) and TP53-induced glycolysis and apoptosis regulator (TIGAR) expression (Fig. 1), therefore, the overall effect of TP53 on cell energy metabolism is to inhibit glycolysis and promote OXPHOS.
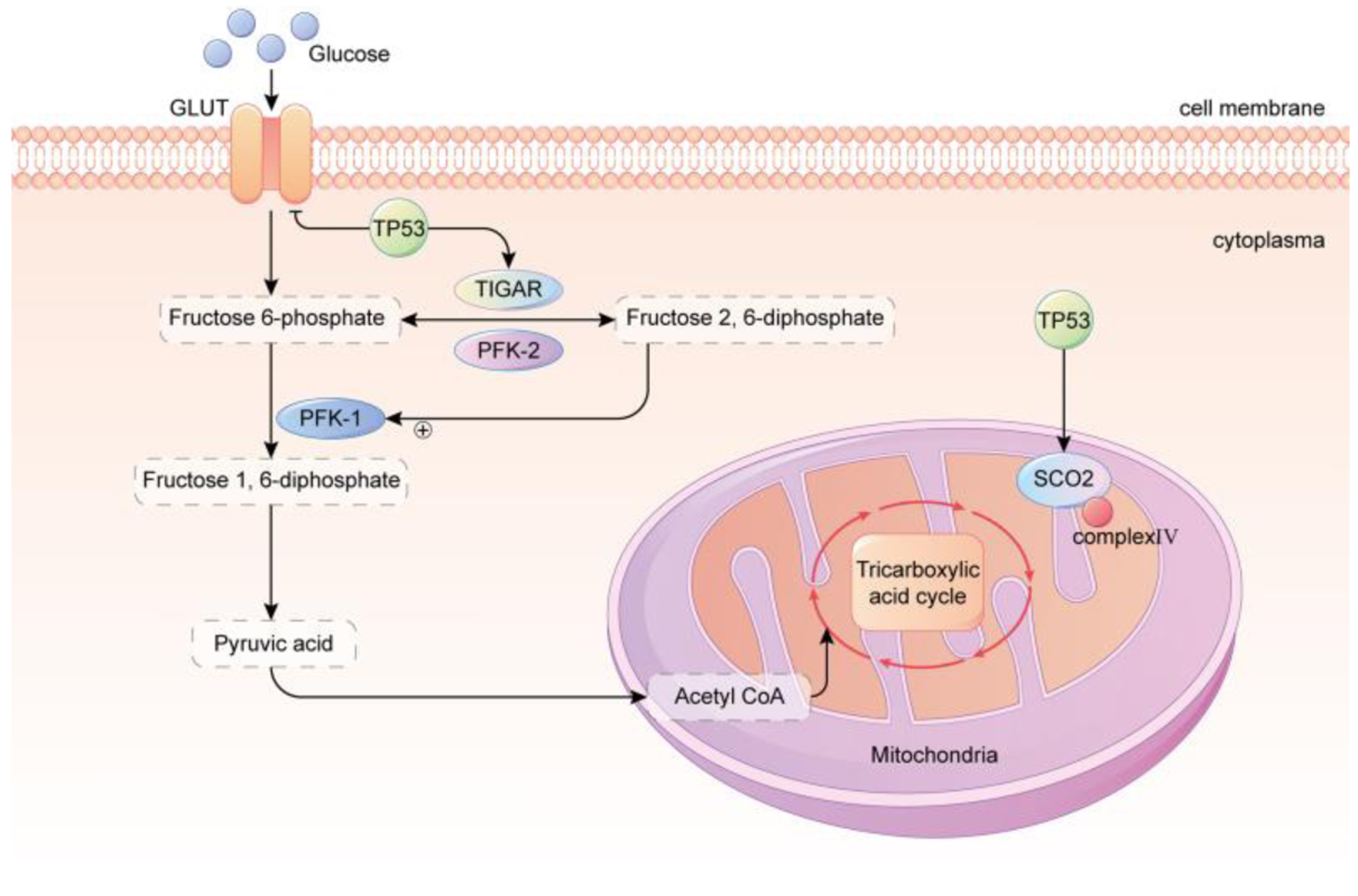 Click for large image | Figure 1. TP53 regulated glycolysis inhibition and OXPHOS promotion. GLUT, which is primarily responsible for transporting glucose, is downregulated by TP53. TP53 upregulates the levels of TIGAR and the mitochondrial respiration chain complex IV subunit SCO2, resulting in a reduction of glycolysis and an enhancement of OXPHOS in the mitochondria. In conclusion, the metabolic phenotype of cancer cells is strongly correlated with the TP53 status. TP53 of wild type possesses robust mitochondrial oxidative phosphorylation and glycolytic reserve capacities, and cells maintain metabolic diversity. In contrast, mutant TP53 cells rely heavily on glycolysis, have a markedly diminished mitochondrial and glycolytic reserve, and operate close to capacity under baseline conditions. GLUT: glucose transporter; TP53: tumor suppressor 53; TIGAR: TP53-induced glycolysis and apoptosis regulator; PFK-1: phosphofructokinase-1; PFK-2: phosphofructokinase-2; SCO2: synthesis of cytochrome c oxidase 2. |
SCO2 participates in the assembly of cytochrome c oxidase (located in the electron transport chain complex IV), which is related to the mitochondrial electron transport chain. Inactivation of TP53 can reduce the activity of SCO2, leading to impaired mitochondrial OXPHOS function [45]. TIGAR reduces fructose-2, 6-bisphosphate (Fru-2, 6-P2) levels, thereby inhibiting glycolysis. Notably, 6-phosphofructokinase 1 (PFK-1) catalyzes the formation of fructose 6-phosphate (Fru-6-P) into fructose 1, 6-diphosphate (Fru-1, 6-P2), the main rate-limiting enzyme in the glycolysis process [46]. Fructose-2, 6-diphosphate is an activator of PFK-1, which regulates the rate of glycolysis (Fig. 1). The inactivation of TP53 can reduce the expression of TIGAR and tilt the energy metabolism of tumor cells towards glycolysis through upregulating PFK-1and Fru-1, 6-P2.
| Clinical Relevance of Tumor Glycolysis and OXPHOS | ▴Top |
Some tumor types have certainly been found to be glycolytic as discussed in above as well as recent research shows. This paves the way for using this enhanced glycolytic flux in tumors for therapeutic intervention. To date several small molecules have been used and are in various pre-clinical and clinical trials for targeting glycolysis in various cancers. There are several classes of these inhibitors, namely inhibitors of glycolytic enzymes (2-deoxy-D-glucose, lonidamine, 3-bromopyruvate (hexokinase), TEPP-46 (PKM2), AZD3965 (monocarboxylate transporter 1)); glucose transport inhibitors (WZB117, BAY-876); LDH inhibitors (oxamate, FX11, quinoline, 3-sulfonamides). Apart from it, glycolysis inhibition can be used in the induction of metabolic stress for making cancer more vulnerable to other cancer therapies, immunotherapy synergy for enhanced anti-cancer immune response, patient stratification on basis of PET scans and adaptive prevention to overcome resistance.
While predominant focus in cancer metabolism has been on glycolysis, the clinical relevance of OXPHOS in tumors is increasingly recognized. The interplay between glycolysis and OXPHOS, as well as the heterogeneity of metabolic profiles among cancer cells, continues to shed light on the diverse roles that OXPHOS plays in cancer progression and treatment response. One of the important challenges of OXPHOS dependent cancers is metabolic plasticity. These cancers activate OXPHOS in tumor cells or adjacent CAFs that gives them adaptability to survive and proliferate in diverse microenvironments and hence cause resistance to anti-glycolytic therapies. Several strategies targeting OXPHOS in cancers are inhibitors of complex 1 of ETC (metformin, IACS-010759, phenformin, BAY 87-2243); ATP synthase inhibitors (oligomycin, rotenone); and mitochondrial uncouplers (2,4-dinitrophenol and carbonyl cyanide p-trifluoromethoxy phenylhydrazone).
Cancer cells are known to undergo a metabolic switch under hypoxia from mitochondrial OXPHOS to glycolysis. The Warburg effect is clinically useful since it is applied in positron emission tomography (PET) scans, where tumor identification is still based on the increased glucose consumption of cancer cells. A study from Oshi et al [47] revealed that in triple negative breast cancer (TNBC), a high glycolysis score was significantly associated with worse patient survival, but not consistently in the estrogen receptor (ER)-positive/human epidermal growth factor receptor 2 (HER2)-negative subtype. However, another finding by Chiche et al [48] showed that the more aggressive diffuse large B-cell lymphoma (DLBCL) subtype has higher OXPHOS and lower glycolysis level, which is different from our well-established notion that the more aggressive tumor cells are glycolytic. Specifically, GAPDH low lymphomas utilize OXPHOS metabolism to activate mTORC1 signaling and glutaminolysis. Consistently, disruptors of OXPHOS metabolism (phenformin) or glutaminolysis (L-asparaginase) could induce cytotoxic responses in GAPDH low B cells and prolong GAPDH low B-cell-lymphoma-bearing mice survival time, but not efficient on GAPDH high B-cell lymphomas.
| Iron Availability Shapes Electron Transfer and OXPHOS | ▴Top |
Iron is a key component of the mitochondrial electron transport chain (OXPHOS site) complexes I, II, and III, and is essential for the formation of iron-sulfur clusters. In addition, a recent study reported that compared with people with normal iron levels, iron-deficient individuals produce less mitochondrial ATP, indicating that the lack of cellular iron directly damages OXPHOS [49]. Using iron-chelating agents or other methods to reduce the iron content in the cell culture environment decreases cell oxygen consumption, which in turn reduces the citrate acid cycle and nicotinamide adenine dinucleotide (NADH) enzymatic activities. Iron deficiency increases the utilization of glucose and glycolysis for energy, while decreasing the oxidation of fatty acids in the respiratory chain of the mitochondria and the production of ATP. In the absence of iron, iron deficiency and oxygen deficiency induce similar metabolic alterations. Iron and oxygen deficiency promotes glucose utilization and glycolysis for energy, increases the production of lactic acid, and inhibits mitochondrial respiratory chain fatty acid OXPHOS (Fig. 2).
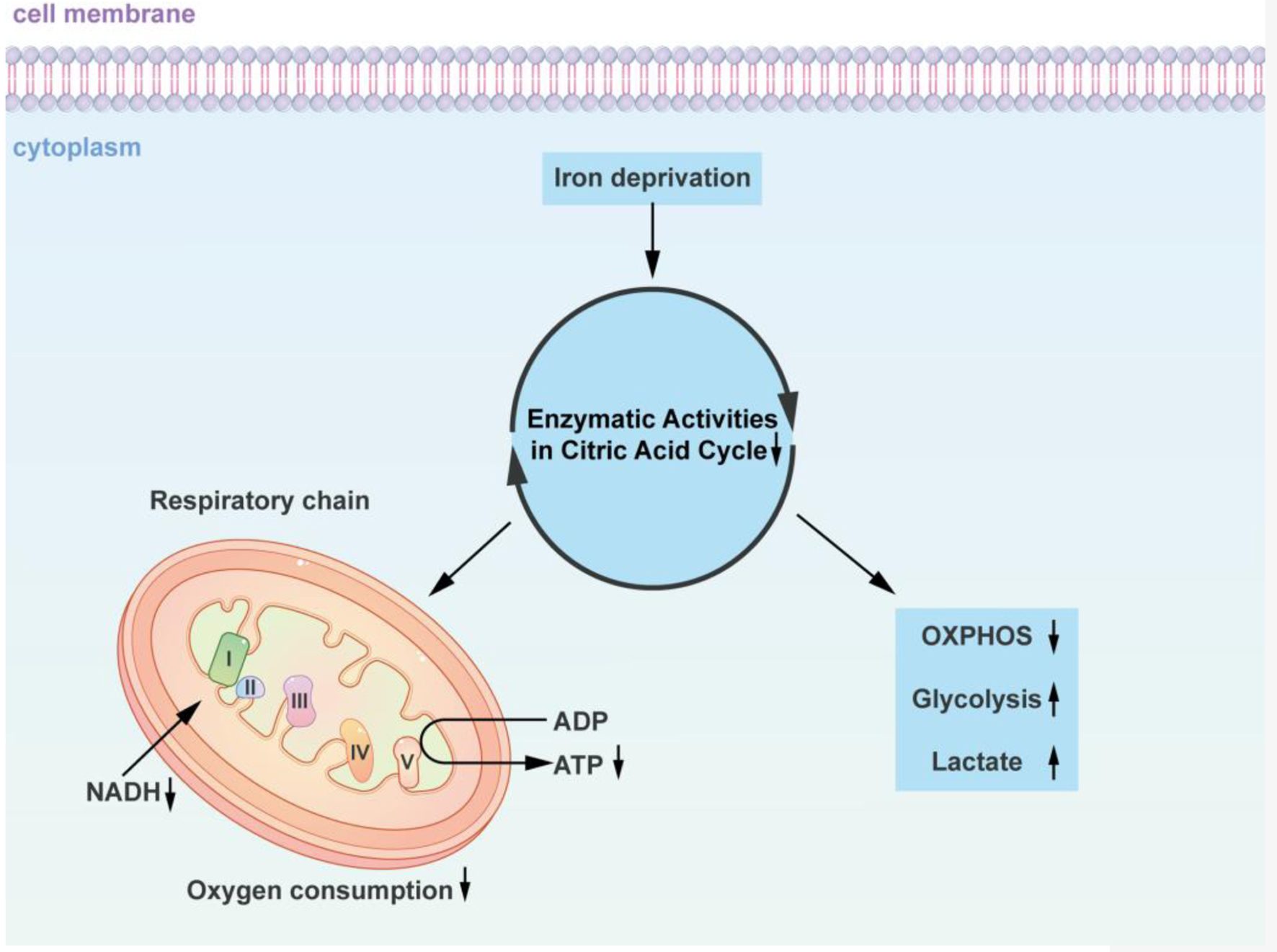 Click for large image | Figure 2. Iron-dependent alterations in cellular energy metabolism: impact on the citric acid cycle and oxidative phosphorylation. The diagram concisely depicts how iron-deficient cells alter the citric acid cycle as well as oxidative phosphorylation. Using iron chelating agents or alternative approaches to reduce the iron content in the cell culture environment decreased the cells’ oxygen consumption, which in turn decreased the citrate acid cycle and NADH enzymatic activities. Iron deficiency promotes glucose and glycolysis utilization for energy, as well as a decline in the oxidation of fatty acids in the respiratory chain of the mitochondria and a decrease in ATP production. Similar metabolic changes are induced by iron deficiency and oxygen deficiency in the absence of iron. Insufficiency in iron and oxygen promotes glucose utilization and glycolysis for energy, increases lactic acid production, and inhibits mitochondrial respiratory chain fatty acid oxidative phosphorylation. NADH: nicotinamide adenine dinucleotide; ADP: adenosine diphosphate; ATP: adenosine triphosphate; OXPHOS: oxidative phosphorylation. |
| Oxygen Availability Regulates Lipid and Sugar Metabolic Pathways | ▴Top |
HIF was identified from nuclear extracts when studying hypoxia-induced erythropoietin (EPO) gene expression in the 1990s [50]. HIF is a basic helix-loop-helix heterodimer, which plays an important role in oxygen balance and adaptation to hypoxia. HIF consists of two subunits, α and β. The α subunit includes HIF-1α, HIF-2α and HIF-3α, and the subtype is regulated by hypoxia signals and the β subunit is stably expressed in the cells [51]. Under normal oxygen conditions, HIF-α is continuously synthesized, but the half-life is less than 5 min and is rapidly degraded. The proline residues in the oxygen-dependent degradation domain (ODDD) of HIF-α are hydroxylated by oxygen and ferrous ion-dependent prolyl hydroxylase (PHD), thereby increasing HIF-α and tumor suppressor protein Von Hippel-Lindau (VHL) affinity, which leads to increased HIF-α protein stability and less degradation.
Iron starvation or hypoxia leads to both upregulation and stabilization of HIF-1α and HIF-2α. HIF-1α stabilization activates the hypoxia-responsive glycolytic genes, such as HK2, GLUT1, and LDH-A, while HIF-2α stabilization deactivates the OXPHOS-related genes, such as Ndufs1, SDHB, and Uqcrfs1 [52]. From this point of view, the shift from OXPHOS to glycolysis for tumor cells can partly be explained by iron starvation or iron deficiency. Inhibition of HIF-1α suppressed the aerobic glycolysis, and inhibition of HIF-2α restored the activities of OXPHOS and mitochondrial complexes I-III [52]. Hypoxia promotes glycogenesis to provide reserves of energy for cells to endure long-term duress. Under conditions of hypoxia, the TCA cycle is inhibited, and Ac-CoA, which is essential for fatty acid synthesis, is primarily supplied by increased glutamine uptake and metabolism. In addition, the OXPHOS catabolism of fatty acids is inhibited, resulting in an increase in the uptake of lipids from the environment. A summary of how the supply of oxygen regulates lipid and sugar metabolic pathways is shown in Figure 3.
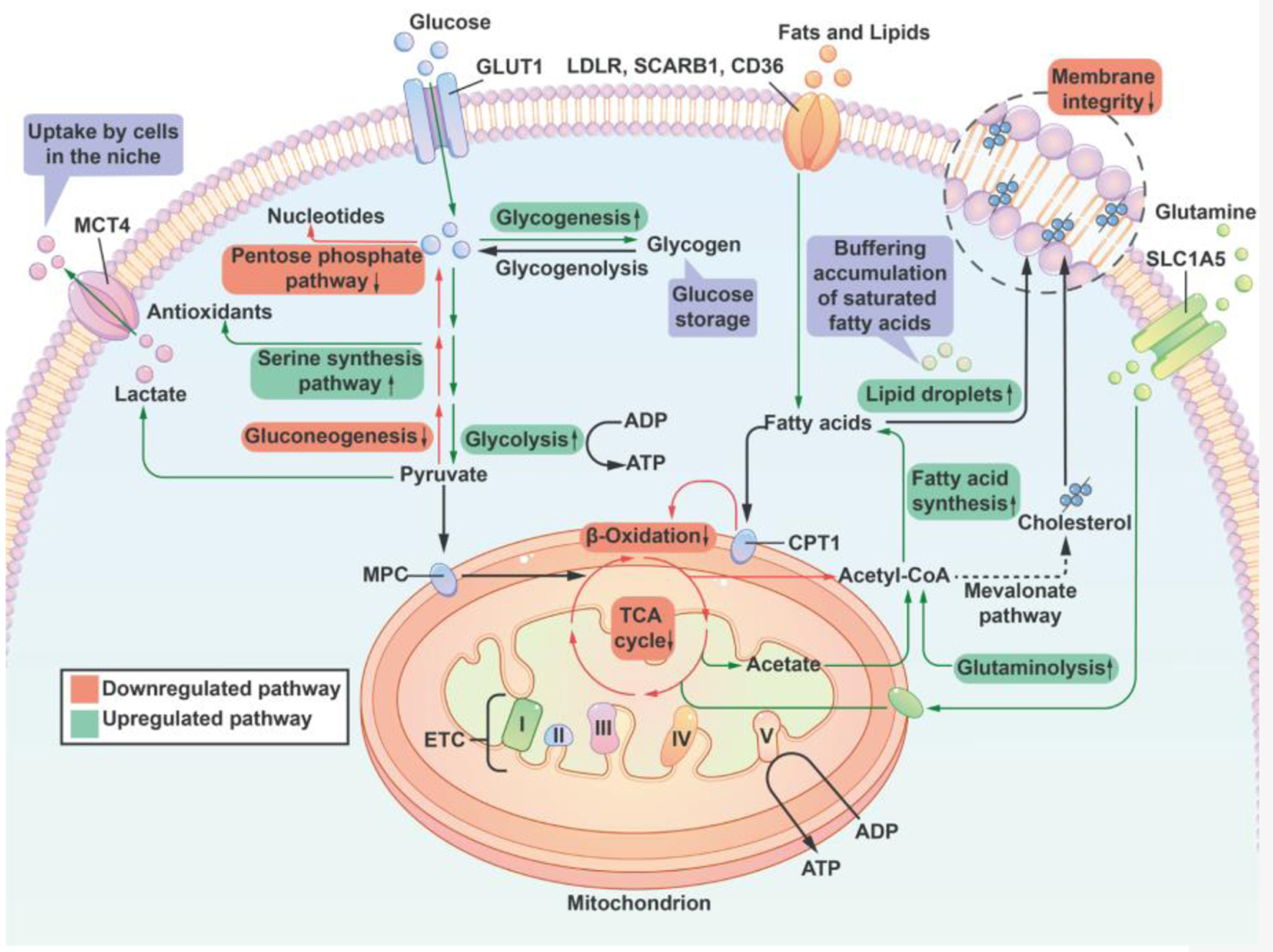 Click for large image | Figure 3. A summary of lipid and sugar metabolic pathways that are regulated by the supply of oxygen. Cells reduce the activity of the pentose phosphate pathway and nucleotide synthesis to compensate for a decrease of antioxidant capacity thereby offering cell protection. Additionally, hypoxia promotes glycogenesis to provide reserves of energy for cells to endure long-term duress. Under conditions of hypoxia, the TCA cycle is inhibited, and Ac-CoA, which is essential for fatty acid synthesis, is primarily supplied by increased glutamine uptake and metabolism. At the same time, the oxidative phosphorylation catabolism of fatty acids is inhibited, resulting in an increase in the uptake of lipids from the environment. Hypoxia inhibits the synthesis of unsaturated fatty acids (a reduced activity of stearoyl-CoA desaturase due to insufficient oxygen). To counteract the potential lipotoxicity caused by the accumulation of saturated fat, cells increased their intake of unsaturated lipids from the external environment and the development of lipid droplets in order to maintain the structural integrity of the cell membrane. Lipid droplets can serve as a buffer for lipid species that are saturated in order to stabilize the cell membrane. I, II, III, IV: respiratory complex I-IV; ETC: electron transport chain; CPT1: carnitine palmitoyl transferase 1; GLUT1: glucose transporter 1; MCT4: monocarboxylic acid transporter 4; LDLR: low-density lipoprotein receptor; MPC: mitochondrial pyruvate carrier; SLC1A5: member of solute carrier family 1 (neutral amino acid transporter) 5; SCARB1: scavenger receptor B1; TCA: tricarboxylic acid; Ac-CoA: Acetyl CoA. |
Cells reduce the activity of the pentose phosphate pathway and nucleotide synthesis to compensate for a decrease in antioxidant capacity thereby offering cell protection. Additionally, hypoxia promotes glycogenesis to provide reserves of energy for cells to endure long-term duress. Under hypoxic conditions, the TCA cycle is inhibited, and Ac-CoA, which is required for fatty acid synthesis, is primarily supplied by an increase in glutamine uptake and metabolism. In addition, the OXPHOS-induced catabolism of fatty acids is inhibited, leading to an increase in lipid absorption from the environment.
| Closing Remarks and Future Directions | ▴Top |
The human genome is coded with the capability to switch between glycolysis and OXPHOS to adapt to different environmental conditions. However, this survival strategy is fully exploited by tumor cells that switch metabolism between glycolysis and OXPHOS to provide energy for survival. Specifically, the shift to glycolysis sustains tumor cell survival under iron and oxygen unavailability, and the relatively insufficient mitochondrial OXPHOS capacity, compared with its fast growth demand. Furthermore, tumors are a heterogeneous disease with very varied genotypes. It is not surprising that the metabolic phenotypes are different. Even within the same tumor site, there are differences in metabolic phenotypes among tumor cells it constitutes. Different cell subgroups form a complementary relationship in metabolism and form a metabolic symbiosis. Notably , during the process of evolution, tumor cells constantly reprogramme metabolism according to environmental changes.
Hypoxia and iron deprivation can both promote tumor cells to glycolysis and sugar utilization, which increased lactic acid production and release into the tumor microenvironment, thus promoting the proliferation of adjacent cells. Fatty acid synthesis is increased under hypoxic conditions, while the fatty acid OXPHOS is decreased. Therefore, targeting the hypoxic and iron-deficient environment of tumors may have potential in tumor treatment and future antitumor research.
Acknowledgments
None to declare.
Financial Disclosure
Rui Wang is founded by China Scholarship Council (202206920039). This research was supported by funds from Natural Science Foundation of Suqian Science and Technology Bureau (K201903, Z2018076, Z2018213 and Z2022065). Jiangsu Association for Science and Technology (JSTJ-2022-004).
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
Rui Wang conceived and wrote the draft, Xiao Wei Jin helped with the figures drawing, Quan Quan Guo and Aashiq Hussain commented and revised the manuscript. Miao Miao Wang helped the revision work. All authors have read and approved the final submission of this manuscript.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
| References | ▴Top |
- Nickelsen K. Otto Warburg's first approach to photosynthesis. Photosynth Res. 2007;92(1):109-120.
doi pubmed - Wisniewski JR, Gizak A, Rakus D. Integrating proteomics and enzyme kinetics reveals tissue-specific types of the glycolytic and gluconeogenic pathways. J Proteome Res. 2015;14(8):3263-3273.
doi pubmed - Nakazawa MS, Keith B, Simon MC. Oxygen availability and metabolic adaptations. Nat Rev Cancer. 2016;16(10):663-673.
doi pubmed pmc - Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292(5516):504-507.
doi pubmed - Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun. 2004;313(3):459-465.
doi pubmed - Suganuma K, Miwa H, Imai N, Shikami M, Gotou M, Goto M, Mizuno S, et al. Energy metabolism of leukemia cells: glycolysis versus oxidative phosphorylation. Leuk Lymphoma. 2010;51(11):2112-2119.
doi pubmed - Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9(6):425-434.
doi pubmed - Moreno-Sanchez R, Rodriguez-Enriquez S, Marin-Hernandez A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274(6):1393-1418.
doi pubmed - Bellance N, Benard G, Furt F, Begueret H, Smolkova K, Passerieux E, Delage JP, et al. Bioenergetics of lung tumors: alteration of mitochondrial biogenesis and respiratory capacity. Int J Biochem Cell Biol. 2009;41(12):2566-2577.
doi pubmed - Jose C, Bellance N, Rossignol R. Choosing between glycolysis and oxidative phosphorylation: a tumor's dilemma? Biochim Biophys Acta. 2011;1807(6):552-561.
doi pubmed - Smolkova K, Plecita-Hlavata L, Bellance N, Benard G, Rossignol R, Jezek P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int J Biochem Cell Biol. 2011;43(7):950-968.
doi pubmed - Herst PM, Berridge MV. Cell surface oxygen consumption: a major contributor to cellular oxygen consumption in glycolytic cancer cell lines. Biochim Biophys Acta. 2007;1767(2):170-177.
doi pubmed - Rodriguez-Enriquez S, Carreno-Fuentes L, Gallardo-Perez JC, Saavedra E, Quezada H, Vega A, Marin-Hernandez A, et al. Oxidative phosphorylation is impaired by prolonged hypoxia in breast and possibly in cervix carcinoma. Int J Biochem Cell Biol. 2010;42(10):1744-1751.
doi pubmed - Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, et al. Ketones and lactate "fuel" tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010;9(17):3506-3514.
doi pubmed pmc - Lim HY, Ho QS, Low J, Choolani M, Wong KP. Respiratory competent mitochondria in human ovarian and peritoneal cancer. Mitochondrion. 2011;11(3):437-443.
doi pubmed - Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8(23):3984-4001.
doi pubmed - Koukourakis MI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer Res. 2006;66(2):632-637.
doi pubmed - Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. 2008;13(6):472-482.
doi pubmed - Brooks GA. Cell-cell and intracellular lactate shuttles. J Physiol. 2009;587(Pt 23):5591-5600.
doi pubmed pmc - Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029-1033.
doi pubmed pmc - Chandra D, Singh KK. Genetic insights into OXPHOS defect and its role in cancer. Biochim Biophys Acta. 2011;1807(6):620-625.
doi pubmed pmc - Owens KM, Kulawiec M, Desouki MM, Vanniarajan A, Singh KK. Impaired OXPHOS complex III in breast cancer. PLoS One. 2011;6(8):e23846.
doi pubmed pmc - Han T, Zhan W, Gan M, Liu F, Yu B, Chin YE, Wang JB. Phosphorylation of glutaminase by PKCepsilon is essential for its enzymatic activity and critically contributes to tumorigenesis. Cell Res. 2018;28(6):655-669.
doi pubmed pmc - Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem. 1979;254(8):2669-2676.
pubmed - DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104(49):19345-19350.
doi pubmed pmc - Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, Miletic H, et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol. 2015;17(12):1556-1568.
doi pubmed pmc - Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15(1):110-121.
doi pubmed pmc - Sandulache VC, Ow TJ, Pickering CR, Frederick MJ, Zhou G, Fokt I, Davis-Malesevich M, et al. Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells. Cancer. 2011;117(13):2926-2938.
doi pubmed pmc - Tong Y, Guo D, Lin SH, Liang J, Yang D, Ma C, Shao F, et al. SUCLA2-coupled regulation of GLS succinylation and activity counteracts oxidative stress in tumor cells. Mol Cell. 2021;81(11):2303-2316.e2308.
doi pubmed - Yang C, Sudderth J, Dang T, Bachoo RM, McDonald JG, DeBerardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009;69(20):7986-7993.
doi pubmed pmc - Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762-765.
doi pubmed pmc - Dejure FR, Royla N, Herold S, Kalb J, Walz S, Ade CP, Mastrobuoni G, et al. The MYC mRNA 3'-UTR couples RNA polymerase II function to glutamine and ribonucleotide levels. EMBO J. 2017;36(13):1854-1868.
doi pubmed pmc - Qing G, Li B, Vu A, Skuli N, Walton ZE, Liu X, Mayes PA, et al. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell. 2012;22(5):631-644.
doi pubmed pmc - Beckner ME, Gobbel GT, Abounader R, Burovic F, Agostino NR, Laterra J, Pollack IF. Glycolytic glioma cells with active glycogen synthase are sensitive to PTEN and inhibitors of PI3K and gluconeogenesis. Lab Invest. 2005;85(12):1457-1470.
doi pubmed - Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008;10(5):611-618.
doi pubmed - Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523.
doi pubmed pmc - Alberghina L, Gaglio D, Gelfi C, Moresco RM, Mauri G, Bertolazzi P, Messa C, et al. Cancer cell growth and survival as a system-level property sustained by enhanced glycolysis and mitochondrial metabolic remodeling. Front Physiol. 2012;3:362.
doi pubmed pmc - Will M, Qin AC, Toy W, Yao Z, Rodrik-Outmezguine V, Schneider C, Huang X, et al. Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling. Cancer Discov. 2014;4(3):334-347.
doi pubmed pmc - Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10(6):594-601.
doi pubmed - Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177-185.
doi pubmed - Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22-35.
doi pubmed pmc - Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105(48):18782-18787.
doi pubmed pmc - Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551(7678):115-118.
doi pubmed pmc - Ma W, Sung HJ, Park JY, Matoba S, Hwang PM. A pivotal role for p53: balancing aerobic respiration and glycolysis. J Bioenerg Biomembr. 2007;39(3):243-246.
doi pubmed - Du W, Amarachintha S, Wilson AF, Pang Q. SCO2 mediates oxidative stress-induced glycolysis to oxidative phosphorylation switch in hematopoietic stem cells. Stem Cells. 2016;34(4):960-971.
doi pubmed pmc - Pilkis SJ, el-Maghrabi MR, Claus TH. Fructose-2,6-bisphosphate in control of hepatic gluconeogenesis. From metabolites to molecular genetics. Diabetes Care. 1990;13(6):582-599.
doi pubmed - Oshi M, Angarita FA, Tokumaru Y, Yan L, Matsuyama R, Endo I, Takabe K. A novel three-gene score as a predictive biomarker for pathologically complete response after neoadjuvant chemotherapy in triple-negative breast cancer. Cancers (Basel). 2021;13(10):2401.
doi pubmed pmc - Chiche J, Reverso-Meinietti J, Mouchotte A, Rubio-Patino C, Mhaidly R, Villa E, Bossowski JP, et al. GAPDH expression predicts the response to R-CHOP, the tumor metabolic status, and the response of DLBCL patients to metabolic inhibitors. Cell Metab. 2019;29(6):1243-1257.e1210.
doi pubmed - Frost JN, Tan TK, Abbas M, Wideman SK, Bonadonna M, Stoffel NU, Wray K, et al. Hepcidin-mediated hypoferremia disrupts immune responses to vaccination and infection. Med. 2021;2(2):164-179.e112.
doi pubmed pmc - Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270(3):1230-1237.
doi pubmed - Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem. 1996;271(30):17771-17778.
doi pubmed - Li H, Liu Y, Shang L, Cai J, Wu J, Zhang W, Pu X, et al. Iron regulatory protein 2 modulates the switch from aerobic glycolysis to oxidative phosphorylation in mouse embryonic fibroblasts. Proc Natl Acad Sci U S A. 2019;116(20):9871-9876.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.