

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 15, Number 2, April 2024, pages 169-180

Sphingosine-1-Phosphate Inhibition Increases Endoplasmic Reticulum Stress to Enhance Oxaliplatin Sensitivity in Pancreatic Cancer
Zachary Gaoa, Harinarayanan Janakiramana, Yang Xiaoa, Sung Wook Kanga, b, c, Jiangling Donga, Jasmine Choia, Besim Ogretmend, Hyun-Sung Leea, b, c, Ernest Ramsay Campa, b, e, f
aMichael E. DeBakey Department of Surgery, Baylor College of Medicine, Houston, TX 77030, USA
bDan L. Duncan Comprehensive Cancer Center, Houston, TX 77030, USA
cSystems Onco-Immunology Laboratory, David J. Sugarbaker Division of Thoracic Surgery, Michael E. DeBakey Department of Surgery, Baylor College of Medicine, Houston, TX 77030, USA
dDepartment of Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC 29425, USA
eMichael E. DeBakey VA Medical Center, Houston, TX 77030, USA
fCorresponding Author: Ernest Ramsay Camp, Michael E. DeBakey Department of Surgery, Baylor College of Medicine, Houston, TX 77030, USA
Manuscript submitted November 14, 2023, accepted January 19, 2024, published online March 21, 2024
Short title: S1P Inhibition in PDAC
doi: https://doi.org/10.14740/wjon1768
| Abstract | ▴Top |
Background: Pancreatic ductal adenocarcinoma (PDAC) is an aggressive cancer resistant to current therapies, including oxaliplatin (Oxa). Growing evidence supports the ability of cancers to harness sphingolipid metabolism for survival. Sphingosine-1-phosphate (S1P) is an anti-apoptotic, pro-survival mediator that can influence cellular functions such as endoplasmic reticulum (ER) stress. We hypothesize that PDAC drives dysregulated sphingolipid metabolism and that S1P inhibition can enhance ER stress to improve therapeutic response to Oxa in PDAC.
Methods: RNA sequencing data of sphingolipid mediators from The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression Project (GTEx) datasets were analyzed. Murine and human PDAC cell lines were treated with small interfering RNA (siRNA) against sphingosine kinase-2 (SPHK2) or ABC294640 (ABC) and incubated with combinations of vehicle control or Oxa. In an orthotopic syngeneic KPC PDAC model, tumors were treated with either vehicle control, Oxa, ABC, or combination therapy.
Results: RNA sequencing analysis revealed multiple significantly differentially expressed sphingolipid mediators (P < 0.05). In vitro, both siRNA knockdown of SPHK2 and ABC sensitized cells to Oxa therapy (P < 0.05), and induced eukaryotic initiation factor 2α (eIF2α) and protein kinase RNA-like endoplasmic reticulum kinase (PERK) phosphorylation, hallmarks of ER stress. In vitro therapy also increased extracellular high mobility group box 1 (HMGB1) release (P < 0.05), necessary for immunogenic cell death (ICD). In vivo combination therapy increased apoptotic markers as well as the intensity of HMGB1 staining compared to control (P < 0.05).
Conclusions: Our evidence suggests that sphingolipid metabolism is dysregulated in PDAC. Furthermore, S1P inhibition can sensitize PDAC to Oxa therapy through increasing ER stress and can potentiate ICD induction. This highlights a potential therapeutic target for chemosensitizing PDAC as well as an adjunct for future chemoimmunotherapy strategies.
Keywords: Sphingosine-1-phosphate; Pancreatic ductal adenocarcinoma; Oxaliplatin; Endoplasmic reticulum stress
| Introduction | ▴Top |
The prognosis for pancreatic ductal adenocarcinoma (PDAC) patients remains poor with 5-year overall survival of less than 10% [1]. Unfortunately, standard conventional chemotherapy regimens including FOLFIRINOX and gemcitabine/nab-paclitaxel provide marginal survival benefit of only months as resistance commonly develops [2, 3]. Similarly, PDAC is resistant to immunotherapeutic strategies either as monotherapy [4] or in combination with chemotherapy [5, 6]. Understanding the underlying mechanisms of chemotherapy resistance is essential to develop strategies to improve outcomes for PDAC patients.
Growing evidence suggests that cancers harness metabolic pathways to promote survival [7-10]. Sphingolipids are bioactive, structural components of the cell membrane involved in cell growth, cellular proliferation, and programmed cell death [11]. A concept of a “rheostat” between pro-apoptotic ceramides and pro-proliferative sphingosine-1-phosphate (S1P) [12, 13] has been well described [14], which may drive a shift between a pro-apoptotic or pro-survival phenotype in cancer cells depending on the balance of these two sphingolipid species [15-17]. Studies support the role of S1P in promoting PDAC cell survival and proliferation [18-22]. Of particular clinical relevance is inhibition of the sphingosine kinase-2 (SPHK2)/S1P axis [23] via the small molecule inhibitor ABC294640 (ABC). ABC is the only drug targeting S1P that has been used in an oncology clinical phase I trial, where it induced a partial treatment response as monotherapy in cholangiocarcinoma and maintained stable disease in other advanced solid tumors [24].
Multiple antineoplastic agents such as cisplatin [25], oxaliplatin (Oxa) [26], and 5-fluorouracil (5-FU) [27] can induce cellular stresses leading to endoplasmic reticulum (ER) stress and activation of the unfolded protein response (UPR) [28]. Continued activation of UPR pathways results in apoptosis [29-32] and immunogenic cell death (ICD) [33, 34]. Interestingly, ER stress and the ceramide/S1P axis intersect at multiple crucial junctions. ER stress and protein kinase R-like endoplasmic reticulum kinase (PERK) activation can induce ceramide generation [35]. Conversely, ER stress can induce S1P synthesis through upregulation of SPHK2 [36] and can utilize S1P as part of downstream signaling mechanisms [37]. Evidence from preclinical cancer models demonstrates that cancer cells can upregulate SPHK2 as a survival mechanism in response to ER stress [38], and increased intracellular S1P can promote cell survival in a PERK-dependent fashion [39].
Together, these data support the rationale for a comprehensive understanding of the role of sphingolipid metabolism in PDAC, as well as investigating the role of SPHK2/S1P as a potential therapy target for PDAC. We hypothesize that PDAC drives dysregulated sphingolipid metabolism and that S1P inhibition can enhance ER stress and ICD to improve therapeutic response to Oxa in PDAC. Thus, the objectives of this study were to first characterize sphingolipid dysfunction via an in silico approach across a large, combined RNA-seq dataset of normal pancreas and PDAC samples. Considering the relationship between S1P and ER stress, we also explored the impact of in vitro antitumoral effect of S1P inhibition via ER stress on ICD induction, both alone and in combination with Oxa. Finally, we investigated the therapeutic effectiveness of S1P inhibition combined with Oxa using an in vivo orthotopic murine syngeneic PDAC model to understand the impact on anti-tumoral immunity.
| Materials and Methods | ▴Top |
Analysis of The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression Project (GTEx) RNA sequencing datasets
Publicly available RNA sequencing data were analyzed using normal pancreatic samples from the GTEx [40] and pancreatic tumor samples from TCGA [41]. The expected RSEM count data from the combined GTEx/TCGA dataset were downloaded from the UCSC Xena platform [42] using UCSCXenaTools v1.4.8 [43]. The RNA sequencing data were back transformed into raw counts and filtered to remove lowly expressed genes and to include only expression data from protein coding genes [44]. An annotation of genes in the sphingolipid metabolic process as well as mediators of the S1P biosynthetic process was curated from Gene Ontology (GO: 0006665) [45] and KEGG (map00600) [46]. The count data for this annotation were converted to log2-counts-per-million values, and unsupervised clustering of these genes was performed in R v4.3.0 using Pearson correlation. Differential gene expression was performed using edgeR v3.42.4 [47] using the quasi-likelihood pipeline with false discovery rate correction at a nominal value of 0.05. The log2 fold change cutoff was set at a nominal value of 0.5.
To understand the clinical association between dysregulated sphingolipid metabolism and clinical outcomes of PDAC, clinical data for PDAC samples from TCGA [41] were analyzed using GEPIA2 [48] for associations between sphingolipid mediator genes and overall survival using Kaplan-Meier estimates.
Institutional Review Board approval is not applicable to this study. This study was conducted in compliance with all the applicable institutional ethical guidelines for the care, welfare, and use of animals (IACUC Protocol #AN-8645).
Cell culture
Human cell lines were obtained directly from the American Type Culture Collection (ATCC). Murine Panc02 cells were obtained from the NCI DCTD Tumor Repository (NCI, Frederick MD) and KPC cells were graciously provided by Dr. Guttridge (Medical University of South Carolina, Charleston, SC). MIAPaCa-2, BxPC-3, HPAC, Capan-2, Panc-1, and AsPC-1 cells were obtained from the ATCC. L3.6pl cells were graciously provided by L. Ellis (MD Anderson, Houston TX). MIAPaCa-2, HPAC, Panc-1, Panc02 and KPC cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (50 IU/mL), streptomycin (50 µg/mL) in a humidified incubator with 5% CO2 at 37 °C. BxPC-3 and AsPC-1 cells were grown in Roswell Park Memorial Institute (RPMI) 1640 media supplemented with 10% FBS, penicillin (50 IU/mL), streptomycin (50 µg/mL) in a humidified incubator with 5% CO2 at 37 °C. Capan-2 cells were grown in McCoys A medium supplemented with 10% FBS, penicillin (50 IU/mL), streptomycin (50 µg/mL) in a humidified incubator with 5% CO2 at 37 °C. All cell lines used in this study were tested for mycoplasma contamination at regular intervals using a PCR-based detection method and submitted for STR authentication at regular intervals.
Reagents
Oxa was purchased from SelleckChem and dissolved in distilled water. ABC was provided by Apogee Biotechnology Corporation (Hummelstown, PA). ABC was dissolved in DMSO for in vitro treatment and in a formula of 46.6% saline, 46.6% PEG 400 and 6.6% Tween-80 for in vivo animal treatment.
SPHK2 knockdown in PDAC cells
SPHK2 knockdown in human PDAC cells was performed by transfection of small interfering RNA (siRNA) which target human SPHK2 gene (targeted sequence: #1, CCCUGAAACUAAACAAGCUUGGUAC and #2, CGUGCUUCCCAUGAUCUCUGAAGCT, 5′ to 3′) with Lipofectamine® 3000 DNA Transfection Reagent (Invitrogen) according to the manufacturer’s protocol. A non-target scrambled siRNA (scrambled sequence: UACAGUUUAUUGAUAUUCAAUAAAG, 5′ to 3′) was used as a negative control of siSPHK2. Forty-eight hours after transfection, cells were added with the indicated treatment in a six-well plate. SPHK2 protein level was determined by immunoblotting with the specific antibody.
Cell viability assay
MTT assay was used to determine cell viability in vitro. Cells were seeded in 96-well plates at 1,000 - 3,000 cells per well for 24 h, followed by indicated treatment for 24 to 72 h. Cells were then incubated with MTT solution (thiazolyl blue tetrazolium bromide, 5 mg/mL) (Tocris Bioscience) at different endpoints at 37 °C for 2 h. After the removal of medium and MTT solution, 100 µL DMSO was added to each well. The absorbance was read at 570/630 nm on a 96-well CLARIOstar (BMG Labtech) plate reader. Each experiment was performed at least in triplicates.
Immunoblotting
Cells were harvested and lysed in TNN buffer (50 mM Tris, 0.25 M NaCl, 5 mM EDTA, and 0.5% Nonidet P-40), and total proteins of tumor samples were extracted in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 0.5% sodium deoxycholate, 1% Nonidet P-40, 0.1% SDS) with protease inhibitor cocktails. After sonication, the protein concentration was measured by BCA assay (Invitrogen). The lysates were boiled in SDS loading buffer, analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel, transferred to the polyvinylidene fluoride (PVDF) membrane (Bio-Rad) and immunoblotted with described antibodies. After imaging of the immunoblots, optical density (OD) was calculated for the detected protein bands using ImageJ by normalizing the target band against the loading control, and then comparing protein bands of interest against a control protein band.
The following antibodies were used for immunoblotting: anti-SPHK2 antibody (#17096-1-AP, Proteintech), anti-phospho-eIF2α antibody (#3597, Cell Signaling), anti-phospho-PERK antibody (#3179, Cell Signaling), anti-PERK antibody (#sc-377400, Santa Cruz), anti-eIF2α antibody (#sc-133132, Santa Cruz), anti-GAPDH antibody (#sc-25778, Santa Cruz), anti-β-actin antibody (#sc-47778, Santa Cruz), ER stress/UPR antibody pack (#NBP2-52746, Novus), anti-rabbit IgG-HRP (#7074, Cell Signaling) and anti-mouse IgG-HRP (#7076, Cell Signaling).
High mobility group box 1 (HMGB1) release detection
The Lumit™ HMGB1 (human) immunoassay kit (Promega) was used to detect HMGB1 released from cultured cell environment undergoing immunogenic cell death. Generally, 10,000 - 15,000 cells were seeded in 96-well plate and followed by indicated treatment next day. The bioluminescence signal was measured on a 96-well CLARIOstar (BMG Labtech) plate reader within 45 min after the addition of Lumit™ substrate. Each experiment was performed at least in triplicates.
Orthotopic KPC in vivo model
This study was conducted in compliance with all the applicable institutional ethical guidelines for the care, welfare, and use of animals. Male C57B1/6 mice of 7 to 8 weeks old were purchased from Jackson Laboratory and housed in a pathogen-free, biohazard barrier facility according to protocols approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine. During the surgery, mice were anesthetized, and the left flank was shaved and sterilized with Betadine. A 1-cm subcostal incision was made through the skin, subcutaneous tissues, and peritoneum. The pancreas was exteriorized, and 0.15 × 106 KPC cells in 0.03 mL PBS (Hyclone) were injected into the pancreatic tail using a sterile 0.5 mL insulin syringe with a 28-gauge needle. Mice were administered pain medication for 3 days post-operatively and sacrificed if evidence of pain or suffering was present.
In vivo animal therapy protocol
After orthotopic injection of KPC cells was performed, 7 to 10 healthy appearing mice were randomized to four treatment groups. Treatment was started 7 days after tumor implantation. Oxa at 3 mg/kg was intraperitoneally injected twice weekly and 50 mg/kg of ABC dissolved in the formula of 46.6% saline, 46.6% PEG 400 and 6.6% Tween-80 was administrated by oral gavage three times per week. The same amount of formula was given as vehicle control of gavage feeding. Mice were monitored for health during the treatment and euthanized 3 weeks after the treatment to measure tumor burden.
Histological analysis
Following sacrifice, tumors were resected, fixed overnight in 10% buffered formalin, set in paraffin blocks, and cut into 5 µm thick sections. Sections were stained with standard hematoxylin and eosin staining. Sections were also stained with Ki67 antibody (1:400 dilution, cat#12202, Cell Signaling), cleaved caspase-3 (CC3) antibody (1:400 dilution, cat#9661, Cell Signaling), HMGB1 antibody (1:1,000 dilution, cat#6893, Cell Signaling), CD3 antibody (1:400 dilution, cat# NB600-1441SS, Novus Biologicals), and CD8 antibody (1:400 dilution, cat#98941s, Cell Signaling). Stained slides were scanned using the EVOS M5000 Imaging System at × 40 magnification after white balance calibration. From each tumor sample, five randomly selected regions chosen by a lab technician blinded to treatment groups were quantified for positively stained nuclei per high power field using QuPath. The intensity of stained nuclei was calculated by thresholding the stained nuclear OD into low, medium, and high intensity staining and converted into an H-score.
Statistical analysis
Statistical analysis was performed using GraphPad PRISM v9.5.0. The unpaired two-tailed t-test or two-way analysis of variance (ANOVA) were used to compare the differences between experimental groups, where appropriate. The false discovery rate was controlled using the two-stage step-up method of Benjamini, Krieger and Yekutieli [49], with a q-value < 0.05 considered statistically significant. Similarly, for multiple comparisons, Tukey’s honest significant difference (HSD) test was used for multiple comparisons in two-way ANOVA, with an adjusted P-value < 0.05 considered statistically significant.
| Results | ▴Top |
Sphingolipid metabolism is dysregulated in PDAC
To evaluate the role of sphingolipid metabolic dysregulation in PDAC, we initially analyzed publicly available RNA sequencing data of normal and pancreatic tumor samples. In total, 167 normal pancreatic tissue samples from GTEx and 183 pancreatic tumor samples from TCGA were included in this analysis. Unsupervised clustering of annotated sphingolipid metabolic genes discriminated between normal pancreatic tissue and PDAC (Fig. 1a). On comparison of PDAC and normal tissue, 100 of the 140 sphingolipid metabolic genes were significantly differentially expressed with a log2 fold change greater or less than 0.5 (Fig. 1b). For example, serine palmitoyltransferase is the initial and also rate-limiting enzyme in global sphingolipid biosynthesis to form 3-ketosphinganine [50]. Two of its isoforms, SPTLC1 and SPTLC2, are significantly upregulated in PDAC. Other S1P biosynthesis pathway genes including ACER1, ACER3, and SPHK1 were significantly upregulated compared to normal pancreatic tissue (Fig. 1b). The immediate downstream receptors of S1P are five G-protein-coupled receptors known as sphingosine-1-phosphate receptors (S1PR1-5) [51]. Four of the five S1P receptors (S1PR1-4) are also significantly upregulated in PDAC compared to normal pancreas. Similarly, a panel of pancreatic cancer cell lines demonstrated strong expression of upstream and downstream S1P mediators (Fig. 2a). Based on the TCGA PDAC patient cohort, multiple significantly overexpressed sphingolipid genes involved in S1P biosynthesis such as KDSR, SPHK1, and SPTLC2 are significantly associated with worsened survival (P < 0.05) (Supplementary Material 1, www.wjon.org). Taken together, these data suggest that S1P biosynthetic pathways (Fig. 2b) are highly dysregulated in PDAC and likely contribute to aggressive biology observed in patients with PDAC.
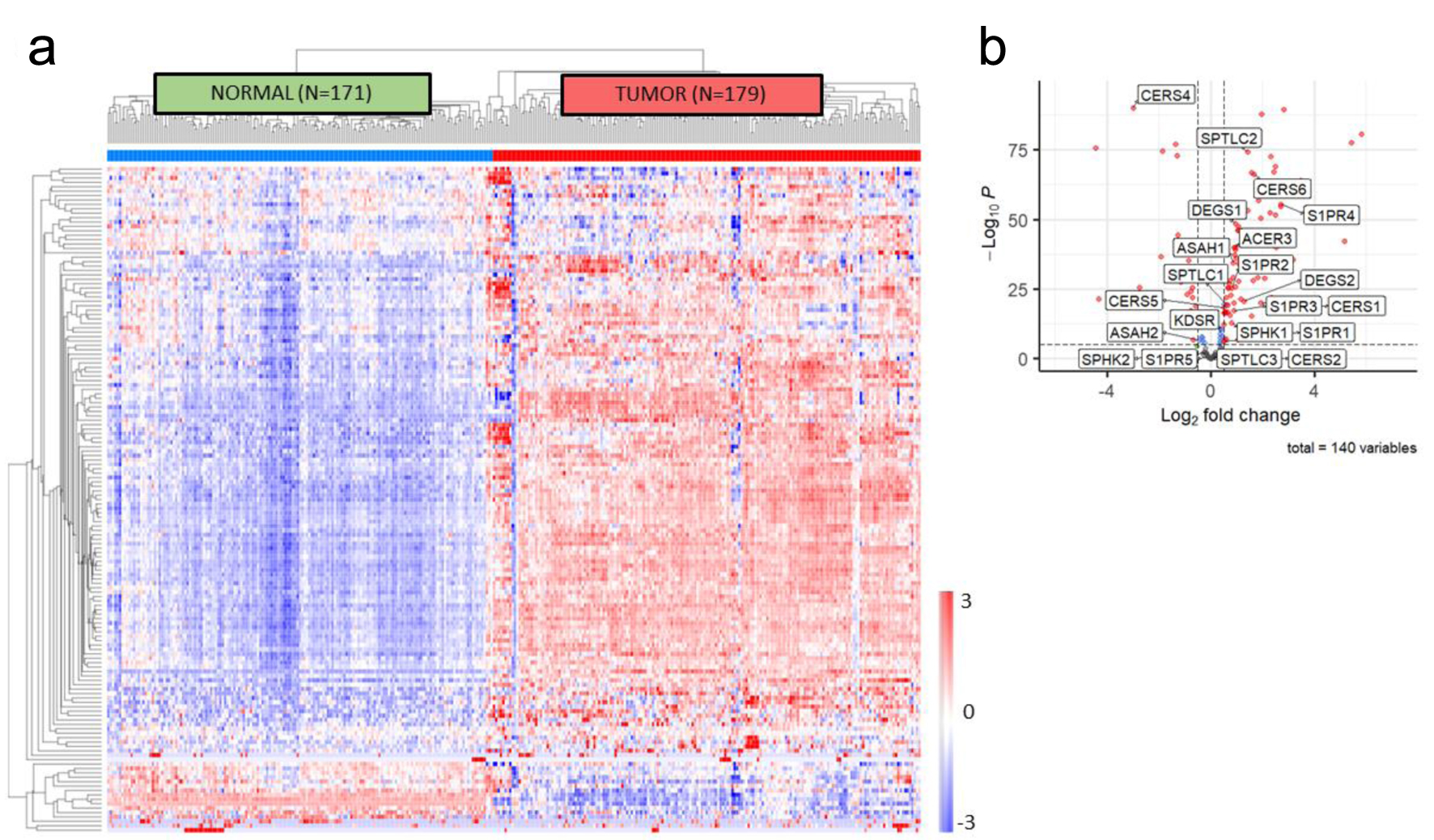 Click for large image | Figure 1. In silico sphingolipid profiling in PDAC. (a) Unsupervised clustering of genes included in the sphingolipid metabolic annotation (GO: 0006665) demonstrates accurate discrimination of normal pancreas tissue from pancreatic tumor tissue using samples from GTEx and TCGA. (b) Volcano plot of differentially expressed genes in the sphingolipid metabolic annotation, with S1P biosynthetic pathway members identified. PDAC: pancreatic ductal adenocarcinoma; S1P: sphingosine-1-phosphate. |
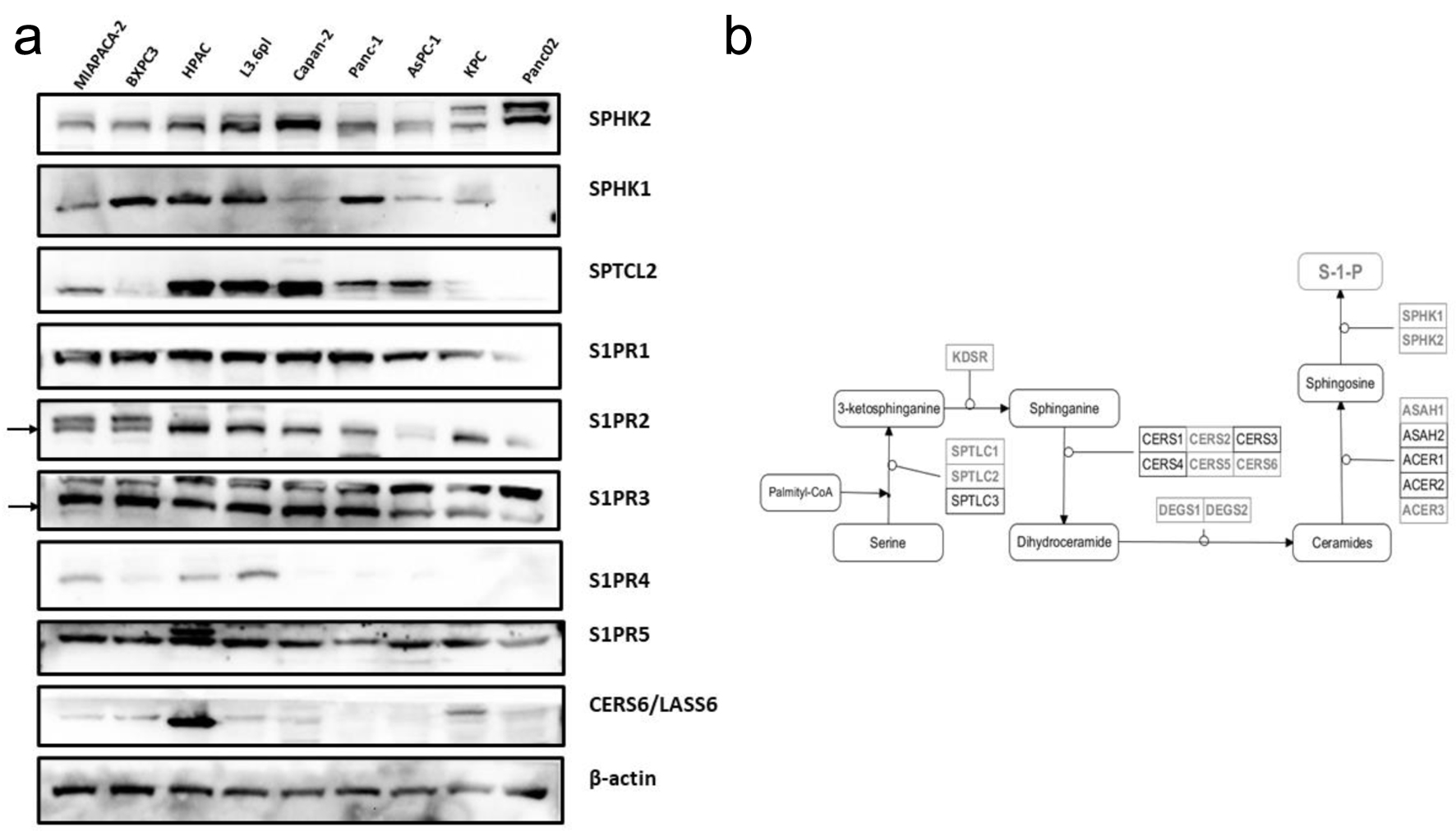 Click for large image | Figure 2. Sphingolipid mediator profiling across human and murine PDAC cell lines. (a) Western blot profiling of upstream and downstream mediators of S1P signaling. (b) Diagram of the biosynthetic pathway of S1P synthesis, with significantly differentially expressed members highlighted. PDAC: pancreatic ductal adenocarcinoma; S1P: sphingosine-1-phosphate. |
S1P signaling inhibition enhances the antitumoral activity of Oxa
Next, we investigated the antitumoral effects of S1P signaling inhibition in PDAC cell lines. Based on cell viability assay, 48-h knockdown of SPHK2 (siSPHK2) demonstrated no significant change in cell viability compared to controls across multiple PDAC cell lines. However, siSPHK2 significantly enhanced Oxa cytotoxicity in multiple cell lines (Fig. 3a). For example, in the MiaPACA-2 cells, the addition of siSPHK2 to Oxa decreased cell viability by 40% compared to cells treated with control scramble siRNA and Oxa (P-value < 0.001). SPHK2 inhibition successfully sensitized PDAC cells to Oxa treatment at both high and low Oxa doses. The effect of SPHK2 inhibition was confirmed using an additional siRNA clone.
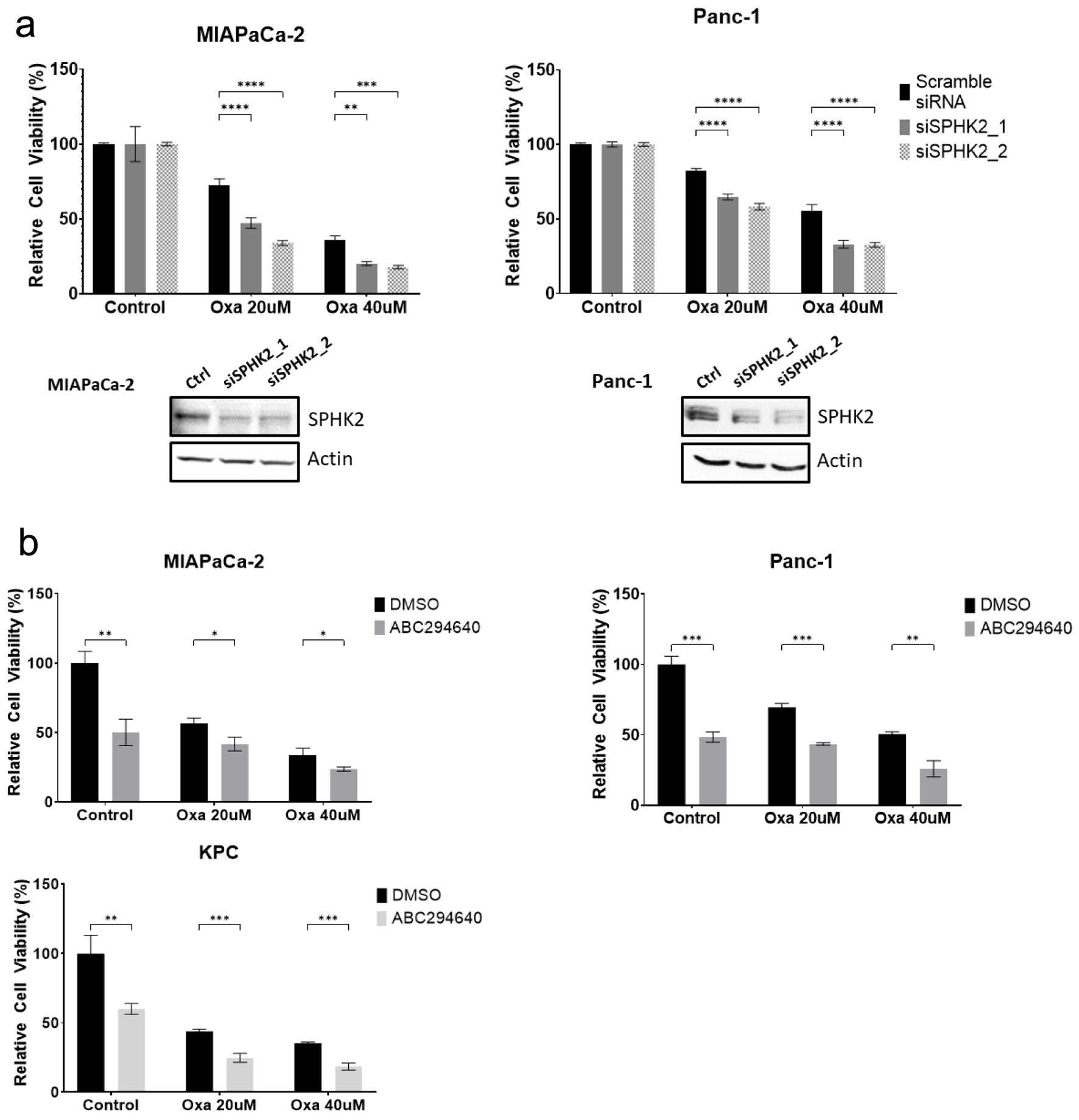 Click for large image | Figure 3. SPHK2 inhibition enhances cytotoxicity of Oxa in PDAC cell lines. (a) After transfection with either siSPHK2 (black bars) or scramble control (gray bars), MiaPaCa-2 and Panc-1 cells were treated with Oxa for 48 h and tested for cell viability based on MTT assay. Western blots confirming effective knockdown of SPHK2 using siRNA clones are displayed below. (b) PDAC cell lines were treated with combination of 20 µM ABC and Oxa for 48 h. Cell viability was assessed by MTT assay. For cell viability assay, data shown represent at least three biological replicates (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). Oxa: oxaliplatin; PDAC: pancreatic ductal adenocarcinoma; S1P: sphingosine-1-phosphate; SPHK2: sphingosine kinase 2. |
To further explore the ability of SPHK2-S1P inhibition to sensitize PDAC cells to Oxa, we tested the combination of ABC, a small molecular inhibitor of SPHK2-S1P signaling, and Oxa. While treatment with ABC alone inhibited cell viability, we observed that the combination of ABC and Oxa increased cancer cell cytotoxicity compared to Oxa alone across multiple PDAC cell lines. In Panc-1 and KPC PDAC cell lines, treatment with ABC decreased cell viability by an additional 20% compared to treatment with 20 µM of Oxa alone (P-value < 0.001, Fig. 3b). This effect was apparent at multiple doses of Oxa, similar to the combination of siSPHK2 with Oxa. Taken together, we demonstrate that therapeutic inhibition of SPHK2-S1P signaling can enhance the antitumoral effects of Oxa.
S1P signaling inhibition enhances Oxa-induced ER stress and ICD in PDAC
Oxa is a well-described agent that drives ER stress [26] and UPR through phosphorylation [52] of the PERK/eukaryotic initiation factor 2α (eIF2α) axis [26], as well as immune activation through ICD [33, 34]. Considering the enhanced cytotoxicity of combination Oxa and S1P inhibition in PDAC, we explored the role of S1P in mediating response to Oxa and its immunogenic effects. In both human and murine PDAC cell lines, Oxa administration induced ER stress and phosphorylation/activation of the PERK/eIF2α pathway (Fig. 4a). siSPHK2 alone mildly activated the ER stress pathway through PERK/eIF2α axis, with a twofold increase in phosphorylated eIF2α expression and phosphorylated PERK expression across both cell lines based on densitometry analysis (Fig. 4a, b). Further, the combination of Oxa and SPHK2 knockdown induced a greater degree of ER stress and UPR activation compared to either treatment alone. For example, in the KPC cell line, combination treatment produced a 1.3-fold increase in phosphorylated PERK expression and a twofold increase in phosphorylated eIF2α expression compared to the cells treated with control scramble siRNA and Oxa. Similar to the results with siSPHK2, ABC (Fig. 4b) increased expression of ER stress mediators and increased the response to Oxa. While either Oxa or ABC treatment alone of KPC cells both increased protein expression of the PERK/eIF2α pathway, combination therapy leads to a 2.6-fold increase in phosphorylated PERK expression and a fivefold increase in phosphorylated eIF2α expression compared to controls. This, along with our previous data demonstrating the combined antitumoral efficacy of S1P signaling inhibition and Oxa, further supports the concept that inhibition of S1P signaling can increase Oxa-induced ER stress in PDAC.
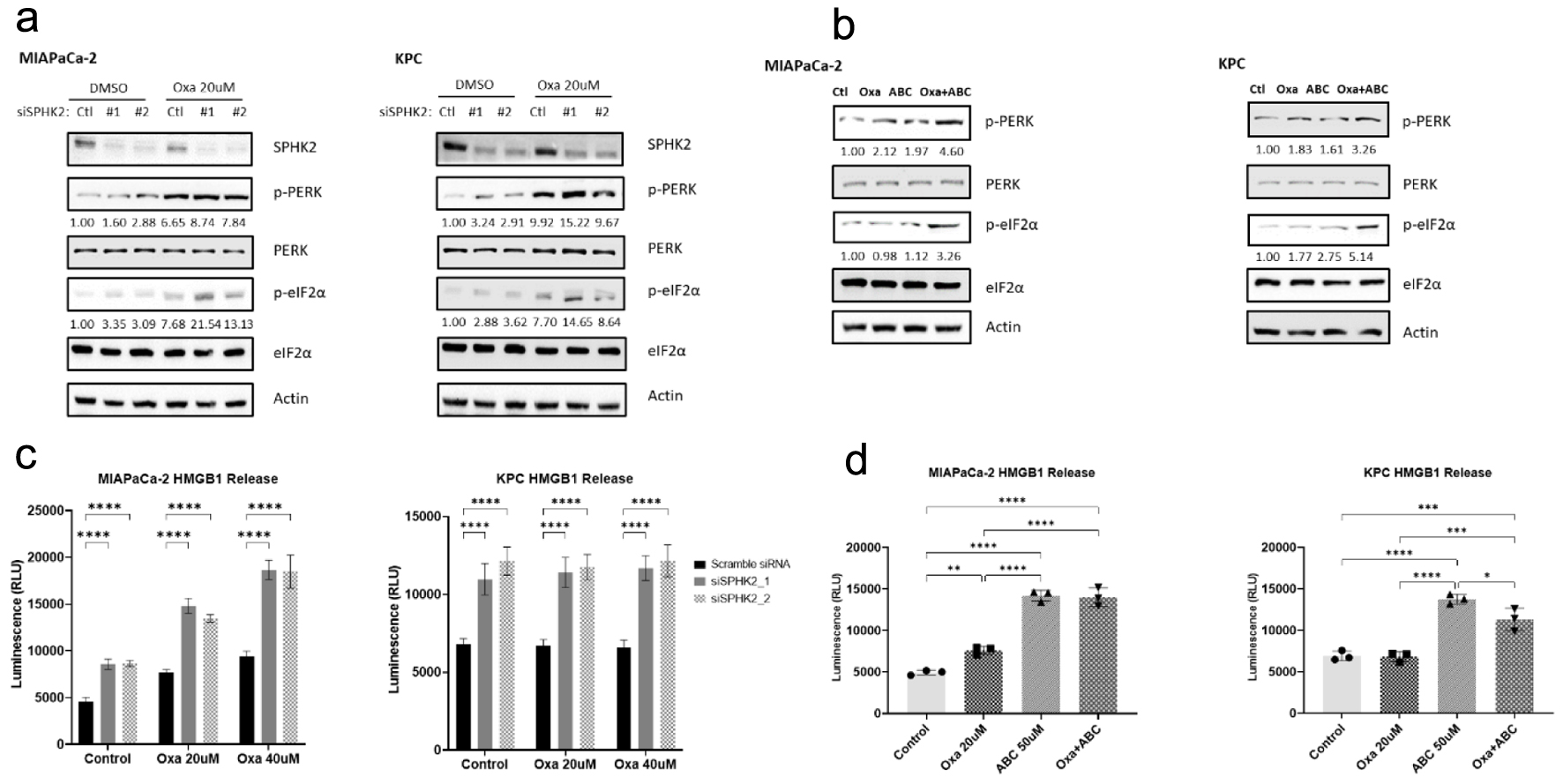 Click for large image | Figure 4. SPHK2 inhibition enhances oxaliplatin-induced ER stress in PDAC cell lines. (a) Loss of SPHK2 increases ER stress through eIF2α and PERK phosphorylation in human and murine cell lines. OD values for p-PERK and p-eIF2α are displayed below the relevant bands. (b) The S1P inhibitor ABC increases ER stress through eIF2α and PERK phosphorylation in human and murine cell lines. OD values for p-PERK and p-eIF2α are displayed below the relevant bands. (c) Loss of SPHK2 and (d) ABC administration increase extracellular HMGB1 release compared to control or Oxa, suggestive of increased ICD induction (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). ABC: ABC294640; ER: endoplasmic reticulum; OD: optical density; Oxa: oxaliplatin; PDAC: pancreatic ductal adenocarcinoma; S1P: sphingosine-1-phosphate; SPHK2: sphingosine kinase 2. |
Considering the close relationship between ER stress and ICD [33, 34, 53, 54], as well as the strong influence of S1P signaling on ER stress, we investigated the ability of S1P to augment ICD. HMGB1 is a key mediator of ICD [55, 56]. Compared with control scramble siRNA, siSPHK2 resulted in a twofold increase in extracellular HMGB1 after 72 h (Fig. 4b, P-value < 0.0001). Experimental results with ABC inhibiting the SPHK2-S1P pathway produce similar results (Fig. 4c). Compared to DMSO control, treatment with ABC produced an almost threefold increase in HMGB1 release in MIAPaCa-2 cells (P-value < 0.0001) and a twofold increase in KPC cells (P-value < 0.0001). ABC administration also led to significant increases in HMGB1 expression compared to Oxa treatment alone in both MIAPaCa-2 and KPC cell lines (P-values < 0.0001). This effect was sustained with combination treatment, as treatment with ABC with Oxa maintained a twofold increase in HMGB1 release compared to Oxa alone across both cell lines (P-values < 0.0001). The increase in extracellular HMGB1 release is consistent across different doses of Oxa treatment, suggesting that S1P inhibition can potentiate the post-apoptotic release of HMGB1 associated with Oxa treatment [57] to stimulate a more robust ICD response in PDAC. Taken together, our evidence supports the concept that S1P signaling inhibition can potentiate ICD induction from conventional chemotherapy.
Combination Oxa with S1P inhibition in an orthotopic PDAC model
Next, we investigated the in vivo antitumor effect of the combination of S1P inhibition inhibitor and Oxa using an orthotopic KPC model (Fig. 5a). Mice with KPC tumors received treatment for 21 days at which time tumors were harvested and analyzed. As expected, Oxa reduced tumor weight compared with vehicle control by 51% (Fig. 5b, P-value = 0.03). The combination of ABC and Oxa reduced mean tumor weight by 17% compared to Oxa, although this was not statistically different (P-value = 0.72). Considering the robust ER stress and ICD signaling in response to combination S1P and Oxa, we further evaluated the ability of ABC to potentiate Oxa effects on proliferation and apoptosis (Fig. 5c, d). Based on immunohistochemical staining, Oxa significantly reduced Ki67 expression compared to the control group (P-value < 0.0001), consistent with the known anti-proliferative effects of Oxa. However, we did not observe significant differences in Ki67 with ABC treatment either as monotherapy compared to control, or with combination treatment compared to Oxa alone (P-values = 0.13 and 0.64, respectively), suggesting that ABC did not exert an in vivo antiproliferative effect. In contrast, the combination of Oxa and ABC significantly increased apoptotic CC3 staining more than twofold compared to control (P-value = 0.025). This effect was not observed with either ABC or Oxa treatment alone, suggesting that ABC can potentiate the therapeutic benefit of Oxa in PDAC.
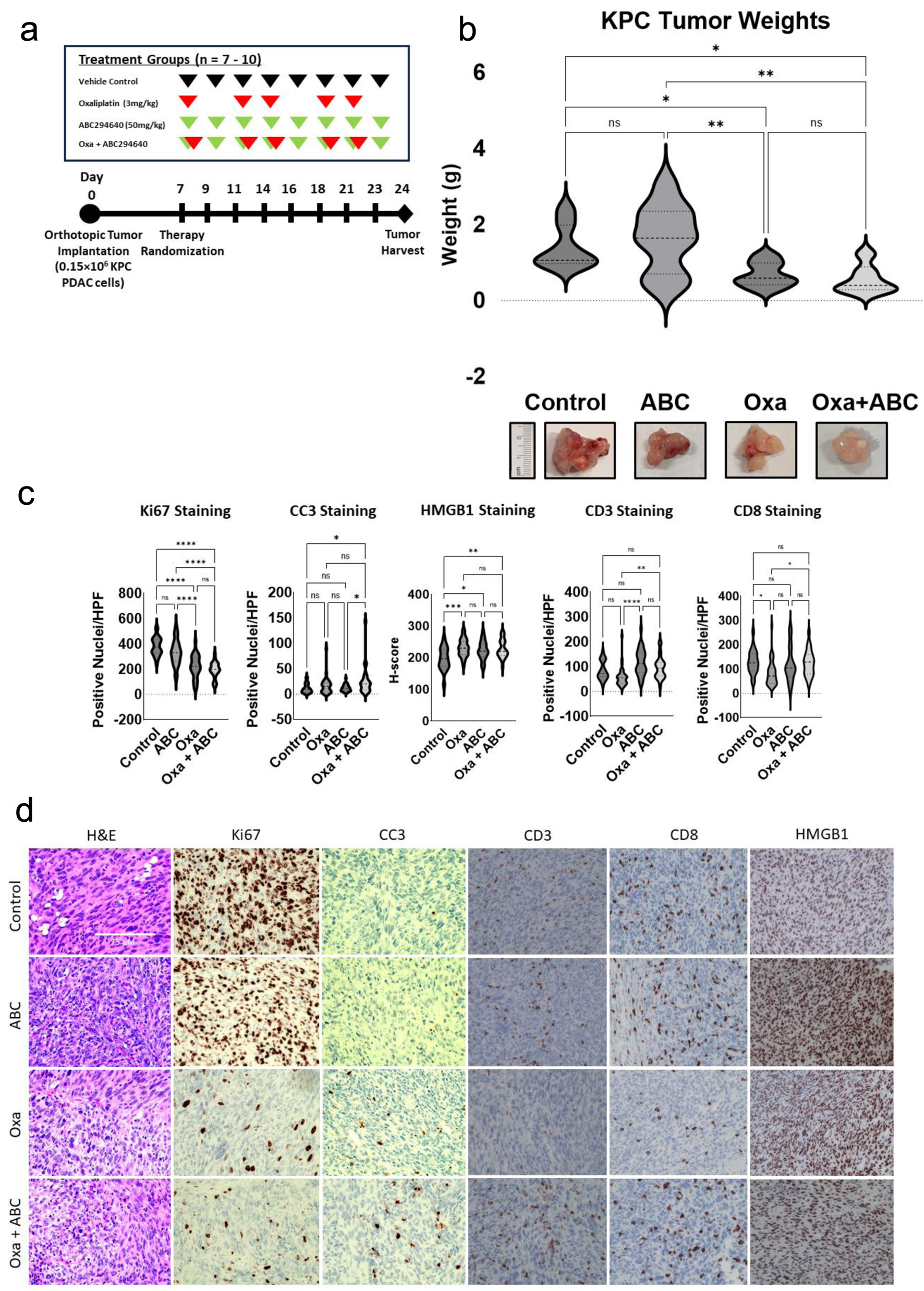 Click for large image | Figure 5. SPHK2-S1P inhibition enhances antitumor activity of Oxa in an orthotopic KPC model. Following treatment for 21 days, orthotopic tumors were harvested and analyzed. (a) In vivo experimental schema. After orthotopic injection of KPC cells, mice were randomized to four treatment groups of 7 - 10 mice each, starting 7 days post-operatively. Oxa at 3 mg/kg (red arrows) was intraperitoneally injected twice weekly and 50 mg/kg of ABC (green arrows) was administrated by oral gavage three times per week. Tumors were harvested 3 weeks after tumor implantation. (b) Analysis of harvested tumor weights by treatment group (*P < 0.05, **P < 0.01). (c) Analysis of IHC stained tumor samples of positively stained nuclei/HPF for Ki67, CC3, CD3, and CD8 between treatment groups. Intensity of HMGB1 staining was quantified by calculating an H-score (*P < 0.05, **P < 0.01). (d) Representative images of IHC stained images of tumors in different treatment groups. ABC: ABC294640; HPF: high power field; IHC: immunohistochemical; Oxa: oxaliplatin; S1P: sphingosine-1-phosphate; SPHK2: sphingosine kinase 2. |
Considering the findings that SphK2 inhibition enhances cellular ER stress and ICD signaling, we explored whether ABC could enhance Oxa-mediated ICD and immune activation in the orthotopic PDAC model (Fig. 5c). Treatment with Oxa, ABC, or combined Oxa and ABC all lead to significantly increased intensity of HMGB1 staining compared to vehicle control (P-values < 0.001, < 0.03, and < 0.01, respectively), although there was no significant difference within these treatment groups. We also noted a trend toward decreased CD3 staining (P-value = 0.13) as well as a significant 33% decrease in CD8 staining (P-value = 0.028) with Oxa monotherapy treatment compared to vehicle control. However, the combined administration of ABC and Oxa led to a robust increase in CD3 (P-value = 0.006) and CD8 (P-value = 0.04) compared to Oxa alone.
| Discussion | ▴Top |
Understanding chemoresistance mechanisms in PDAC is crucial to improving therapeutic strategies to impact patient outcomes [2, 3]. Cancers, including PDAC, effectively harness metabolic mediators such as sphingolipids to promote survival, metastasis, and therapy resistance [8, 14, 21, 22]. Considering the close relationship between SPHK2-S1P signaling and ER stress, we hypothesized that PDAC drives dysregulated sphingolipid metabolism and that S1P inhibition can enhance ER stress and ICD to improve therapeutic and immunologic response to Oxa in PDAC.
In the present study, we demonstrated sphingolipid metabolism is highly dysregulated in PDAC compared to normal pancreatic tissue. The majority of genes in the S1P biosynthetic pathways and immediate downstream S1P receptors are significantly upregulated in PDAC, while key members such as KDSR, SPHK1, and SPTLC2 are associated with worsened survival. Combined with previous studies that observed higher levels of S1P in tumor [18] and its associations with PDAC tumor growth [19] and carcinomatosis [20], our evidence suggests that the ceramide-S1P rheostat [14] is shifted to a pro-survival phenotype, and strongly implicates S1P biosynthetic and signaling pathways as mediators of the aggressive tumor biology of PDAC.
In regard to the relationship between ER stress and SPHK2, we further explored one aspect of sphingolipid and S1P signaling in PDAC by determining its function in mediating chemoresistance. Our experimental data demonstrate that targeting the SPHK2-S1P axis through either siRNA knockdown or administration of the small-molecule inhibitor ABC results in increased chemosensitivity to Oxa at multiple doses and across multiple human and murine PDAC cell lines. We also demonstrate that while both Oxa and S1P inhibition increase ER stress and subsequent phosphorylation of the PERK/eIF2α pathway, the combination of the two therapies induces a greater degree of phosphorylation compared to either treatment alone. Taken together, our data suggest that physiologically, SPHK2-S1P signaling serves to blunt Oxa-induced ER stress and UPR activation. PDAC possesses a hypoxic, nutrient-deprived microenvironment [58] and exhibits a basal amount of ER stress [59] in response to its disadvantageous microenvironmental milieu. However, continual accumulation of misfolded proteins and persistent ER stress without resolution leads to apoptotic cell death [29-32]. Inhibition of the SPHK2-S1P axis may further increase basal and Oxa-induced ER stress, leading to an enhanced pro-apoptotic state as observed in the in vivo experiment. These findings are consistent with similar cancer investigations that have studied the potentiation of Oxa by inducing greater ER stress [60, 61]. The increase in in vivo apoptotic markers in our PDAC tumors is also encouraging and suggests a chemosensitizing role for SPHK2/S1P inhibition.
In addition to chemoresistance, PDAC is plagued by resistance to immunotherapy [4-6, 58]. Induction of ICD has been explored as a potential adjunctive therapy strategy [62-64], and multiple drugs have been investigated but not yet clinically utilized for their ability to induce ICD [65], including Oxa [66]. The potential for S1P inhibition as an immune adjunct is attractive, as the successes seen with immune remodeling from combination in other malignancies such as lung and breast cancer [67, 68] have not been reproduced in PDAC clinical trials [5, 6]. Compared to Oxa alone, in vitro SPHK2 knockdown and ABC administration result in a higher degree of phosphorylation of eIF2α and induce significantly higher levels of extracellular HMGB1 release, both hallmarks of ICD [34, 65]. Our in vivo results show a significant increase in the intensity of HMGB1 staining along with increased CD3 and CD8 marker expression in our tumors treated with ABC and Oxa. We also noted a significant decrease in CD8 staining with Oxa monotherapy compared to vehicle control, which was ameliorated with combination treatment with Oxa and ABC. Oxa administration has been reported to increase the proportion of CD8 T cells in tumors [69-71]. However, in vivo Oxa models have also reported hematological toxicity and suppression of CD8 T cell populations at doses similar to those used in our experiment [72], suggesting that dose and timing optimization is important in designing future chemoimmunotherapy strategies. It is interesting to note that our combination therapy group with Oxa and ABC did not experience this reduction in CD8 staining. These data suggest that ABC may have a protective effect on CD8 tumor immune infiltrates, although the mechanism behind this is not yet elucidated and requires further experiments. Taken together with the increase in apoptotic marker expression seen in the tumors treated with combination therapy, our data argue for a role in SPHK2-S1P signaling inhibition in increasing Oxa-induced ICD and immune activation in PDAC tumors. ICD inducers such as Oxa have also demonstrated the ability to increase PD-1 and PD-L1 expression [66] as well as improved outcomes in combination with immunotherapy in solid tumors [73, 74]. S1P inhibition represents an appealing strategy to potentiate Oxa-induced ICD and may also serve as an immune adjunct to support future chemoimmunotherapy combinations in PDAC.
Conclusions
Our work highlights the significant dysregulation of sphingolipids in PDAC. Furthermore, our evidence implicates sphingolipid metabolism and S1P signaling in mediating chemoresistance to Oxa, as well as highlighting a potential novel therapeutic target for chemosensitizing PDAC. These findings also strongly support future efforts to explore the downstream immune modulatory effects of ER stress and ICD induction following therapeutic targeting of S1P. Finally, the immune activating properties of targeting S1P alone or with chemotherapy point to an exciting strategy to boost chemoimmunotherapeutic strategies for PDAC.
| Supplementary Material | ▴Top |
Suppl 1. Differential expression of selected individual sphingolipid mediators with KRAS mutation and survival.
Acknowledgments
This work was supported in part by Merit Review Award #I01 CX001880-01A1 from the United States (US) Department of Veterans Affairs Biomedical Laboratory Research and Development Program (ERC), and by an award from the National Pancreatic Cancer Foundation. This was also supported by the Dan L Duncan Comprehensive Cancer Center Bench to Bedside Research pilot grant by (Camp & Lee: P30 CA125123).
Financial Disclosure
This work was supported in part by Merit Review Award #I01 CX001880-01A1 from the United States (US) Department of Veterans Affairs Biomedical Laboratory Research and Development Program (ERC), and by an award from the National Pancreatic Cancer Foundation.
Conflict of Interest
The authors declare no potential conflict of interest.
Informed Consent
Not applicable.
Author Contributions
ZG: conceptualization, methodology, formal analysis, investigation, writing - original draft preparation, review, and editing, visualization; HJ: methodology, investigation, writing - review and editing; YX: methodology, investigation, writing - review and editing; SWK: investigation, writing - review and editing; JD: methodology, investigation, writing - review and editing; JC: methodology, investigation, writing - review and editing; BO: writing - review and editing; HSL: writing - review and editing, ERC: conceptualization, resources, supervision, project administration, writing - review and editing.
Data Availability
The data supporting the findings of this study are in whole or part based upon data generated by the TCGA Research Network and can be accessed at https://www.cancer.gov/tcga.
Abbreviations
ABC: ABC294640; ICD: immunogenic cell death; OD: optical density; Oxa: oxaliplatin; PDAC: pancreatic ductal adenocarcinoma; S1P: sphingosine-1-phosphate; SPHK2: sphingosine kinase 2
| References | ▴Top |
- Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17-48.
doi pubmed - Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691-1703.
doi pubmed pmc - Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul JL, Chone L, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med. 2018;379(25):2395-2406.
doi pubmed - Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33(8):828-833.
doi pubmed pmc - Mohindra NA, Kircher SM, Nimeiri HS, Benson AB, Rademaker A, Alonso E, Blatner N, et al. Results of the phase Ib study of ipilimumab and gemcitabine for advanced pancreas cancer. Journal of Clinical Oncology. 2015;33(15_suppl):e15281.
doi - Wainberg ZA, Hochster HS, Kim EJ, George B, Kaylan A, Chiorean EG, Waterhouse DM, et al. Open-label, Phase I study of nivolumab combined with nab-paclitaxel plus gemcitabine in advanced pancreatic cancer. Clin Cancer Res. 2020;26(18):4814-4822.
doi pubmed - Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14(1):11-31.
doi pubmed - Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer. 2018;18(1):33-50.
doi pubmed pmc - Schiliro C, Firestein BL. Mechanisms of metabolic reprogramming in cancer cells supporting enhanced growth and proliferation. Cells. 2021;10(5):1056.
doi pubmed pmc - Luengo A, Gui DY, Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem Biol. 2017;24(9):1161-1180.
doi pubmed pmc - Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004;4(8):604-616.
doi pubmed - Giussani P, Prinetti A, Tringali C. The role of sphingolipids in cancer immunotherapy. Int J Mol Sci. 2021;22(12):6492.
doi pubmed pmc - Janneh AH, Ogretmen B. Targeting sphingolipid metabolism as a therapeutic strategy in cancer treatment. Cancers (Basel). 2022;14(9):2183.
doi pubmed pmc - Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10(7):489-503.
doi pubmed - Schnitzer SE, Weigert A, Zhou J, Brune B. Hypoxia enhances sphingosine kinase 2 activity and provokes sphingosine-1-phosphate-mediated chemoresistance in A549 lung cancer cells. Mol Cancer Res. 2009;7(3):393-401.
doi pubmed - Bonhoure E, Pchejetski D, Aouali N, Morjani H, Levade T, Kohama T, Cuvillier O. Overcoming MDR-associated chemoresistance in HL-60 acute myeloid leukemia cells by targeting sphingosine kinase-1. Leukemia. 2006;20(1):95-102.
doi pubmed - Lifshitz V, Priceman SJ, Li W, Cherryholmes G, Lee H, Makovski-Silverstein A, Borriello L, et al. Sphingosine-1-phosphate receptor-1 promotes environment-mediated and acquired chemoresistance. Mol Cancer Ther. 2017;16(11):2516-2527.
doi pubmed pmc - Yuza K, Nakajima M, Nagahashi M, Tsuchida J, Hirose Y, Miura K, Tajima Y, et al. Different roles of sphingosine kinase 1 and 2 in pancreatic cancer progression. J Surg Res. 2018;232:186-194.
doi pubmed - Bi Y, Li J, Ji B, Kang N, Yang L, Simonetto DA, Kwon JH, et al. Sphingosine-1-phosphate mediates a reciprocal signaling pathway between stellate cells and cancer cells that promotes pancreatic cancer growth. Am J Pathol. 2014;184(10):2791-2802.
doi pubmed pmc - Aoki H, Aoki M, Katsuta E, Ramanathan R, Idowu MO, Spiegel S, Takabe K. Host sphingosine kinase 1 worsens pancreatic cancer peritoneal carcinomatosis. J Surg Res. 2016;205(2):510-517.
doi pubmed pmc - Shen Y, Wang X, Xia W, Wang C, Cai M, Xie H, Zhou L, et al. Antiproliferative and overadditive effects of rapamycin and FTY720 in pancreatic cancer cells in vitro. Transplant Proc. 2008;40(5):1727-1733.
doi pubmed - Shen Y, Cai M, Xia W, Liu J, Zhang Q, Xie H, Wang C, et al. FTY720, a synthetic compound from Isaria sinclairii, inhibits proliferation and induces apoptosis in pancreatic cancer cells. Cancer Lett. 2007;254(2):288-297.
doi pubmed - Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22(1):50-60.
doi pubmed pmc - Britten CD, Garrett-Mayer E, Chin SH, Shirai K, Ogretmen B, Bentz TA, Brisendine A, et al. A phase I study of ABC294640, a first-in-class sphingosine kinase-2 inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2017;23(16):4642-4650.
doi pubmed pmc - Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem. 2003;278(11):9100-9106.
doi pubmed - Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, Durchschlag M, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28(5):578-590.
doi pubmed pmc - Yadunandam AK, Yoon JS, Seong YA, Oh CW, Kim GD. Prospective impact of 5-FU in the induction of endoplasmic reticulum stress, modulation of GRP78 expression and autophagy in Sk-Hep1 cells. Int J Oncol. 2012;41(3):1036-1042.
doi pubmed - Oakes SA. Endoplasmic reticulum stress signaling in cancer cells. Am J Pathol. 2020;190(5):934-946.
doi pubmed pmc - Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15(5):481-490.
doi pubmed pmc - McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21(4):1249-1259.
doi pubmed pmc - Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18(24):3066-3077.
doi pubmed pmc - Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, Bredesen DE, et al. Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem. 2002;277(24):21836-21842.
doi pubmed - Kepp O, Menger L, Vacchelli E, Locher C, Adjemian S, Yamazaki T, Martins I, et al. Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev. 2013;24(4):311-318.
doi pubmed - Kepp O, Semeraro M, Bravo-San Pedro JM, Bloy N, Buque A, Huang X, Zhou H, et al. eIF2alpha phosphorylation as a biomarker of immunogenic cell death. Semin Cancer Biol. 2015;33:86-92.
doi pubmed - Yacoub A, Hamed HA, Allegood J, Mitchell C, Spiegel S, Lesniak MS, Ogretmen B, et al. PERK-dependent regulation of ceramide synthase 6 and thioredoxin play a key role in mda-7/IL-24-induced killing of primary human glioblastoma multiforme cells. Cancer Res. 2010;70(3):1120-1129.
doi pubmed pmc - Lee SY, Hong IK, Kim BR, Shim SM, Sung Lee J, Lee HY, Soo Choi C, et al. Activation of sphingosine kinase 2 by endoplasmic reticulum stress ameliorates hepatic steatosis and insulin resistance in mice. Hepatology. 2015;62(1):135-146.
doi pubmed - Park K, Ikushiro H, Seo HS, Shin KO, Kim YI, Kim JY, Lee YM, et al. ER stress stimulates production of the key antimicrobial peptide, cathelicidin, by forming a previously unidentified intracellular S1P signaling complex. Proc Natl Acad Sci U S A. 2016;113(10):E1334-1342.
doi pubmed pmc - Mizutani N, Omori Y, Tanaka K, Ito H, Takagi A, Kojima T, Nakatochi M, et al. Increased SPHK2 transcription of human colon cancer cells in serum-depleted culture: the involvement of CREB transcription factor. J Cell Biochem. 2015;116(10):2227-2238.
doi pubmed - Lepine S, Allegood JC, Park M, Dent P, Milstien S, Spiegel S. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ. 2011;18(2):350-361.
doi pubmed pmc - Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648-660.
doi pubmed pmc - Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45(10):1113-1120.
doi pubmed pmc - Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A, Banerjee A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38(6):675-678.
doi pubmed pmc - Wang S, Liu X. The UCSCXenaTools R package: a toolkit for accessing genomics data from UCSC Xena platform, from cancer multi-omics to single-cell RNA-seq. Journal of Open Source Software. 2019;4(40):1627.
- Piovesan A, Antonaros F, Vitale L, Strippoli P, Pelleri MC, Caracausi M. Human protein-coding genes and gene feature statistics in 2019. BMC Res Notes. 2019;12(1):315.
doi pubmed pmc - Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25-29.
doi pubmed pmc - Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27-30.
doi pubmed pmc - Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139-140.
doi pubmed pmc - Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47(W1):W556-W560.
doi pubmed pmc - Benjamini Y, Krieger AM, Yekutieli D. Adaptive linear step-up procedures that control the false discovery rate. Biometrika. 2006;93(3):491-507.
- Merrill AH, Jr. Characterization of serine palmitoyltransferase activity in Chinese hamster ovary cells. Biochim Biophys Acta. 1983;754(3):284-291.
doi pubmed - Kihara Y, Maceyka M, Spiegel S, Chun J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br J Pharmacol. 2014;171(15):3575-3594.
doi pubmed pmc - Fels DR, Koumenis C. The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol Ther. 2006;5(7):723-728.
doi pubmed - Sagar V, Vatapalli R, Lysy B, Pamarthy S, Anker JF, Rodriguez Y, Han H, et al. EPHB4 inhibition activates ER stress to promote immunogenic cell death of prostate cancer cells. Cell Death Dis. 2019;10(11):801.
doi pubmed pmc - Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12(12):860-875.
doi pubmed - Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050-1059.
doi pubmed - Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, Mariette C, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220:47-59.
doi pubmed - Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29(4):482-491.
doi pubmed - Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, Bastidas AJ, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys. 2000;48(4):919-922.
doi pubmed - Kong B, Cheng T, Wu W, Regel I, Raulefs S, Friess H, Erkan M, et al. Hypoxia-induced endoplasmic reticulum stress characterizes a necrotic phenotype of pancreatic cancer. Oncotarget. 2015;6(31):32154-32160.
doi pubmed pmc - Lei Y, He L, Yan C, Wang Y, Lv G. PERK activation by CCT020312 chemosensitizes colorectal cancer through inducing apoptosis regulated by ER stress. Biochem Biophys Res Commun. 2021;557:316-322.
doi pubmed - Liu X, Wu B, Chen H, Sun H, Guo X, Sun T, Zhou D, et al. Intense endoplasmic reticulum stress (ERS) / IRE1alpha enhanced Oxaliplatin efficacy by decreased ABCC10 in colorectal cancer cells. BMC Cancer. 2022;22(1):1369.
doi pubmed pmc - Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17(2):97-111.
doi pubmed - Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, Breckpot K, et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol. 2015;6:588.
doi pubmed pmc - Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51-72.
doi pubmed - Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3(9):e955691.
doi pubmed pmc - Zhao X, Yang K, Zhao R, Ji T, Wang X, Yang X, Zhang Y, et al. Inducing enhanced immunogenic cell death with nanocarrier-based drug delivery systems for pancreatic cancer therapy. Biomaterials. 2016;102:187-197.
doi pubmed - Gandhi L, Rodriguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378(22):2078-2092.
doi pubmed - Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379(22):2108-2121.
doi pubmed - Stojanovska V, Prakash M, McQuade R, Fraser S, Apostolopoulos V, Sakkal S, Nurgali K. Oxaliplatin treatment alters systemic immune responses. Biomed Res Int. 2019;2019:4650695.
doi pubmed pmc - Zhou J, Yang T, Liu L, Lu B. Chemotherapy oxaliplatin sensitizes prostate cancer to immune checkpoint blockade therapies via stimulating tumor immunogenicity. Mol Med Rep. 2017;16(3):2868-2874.
doi pubmed - Michelakos T, Cai L, Villani V, Sabbatino F, Kontos F, Fernandez-Del Castillo C, Yamada T, et al. Tumor microenvironment immune response in pancreatic ductal adenocarcinoma patients treated with neoadjuvant therapy. J Natl Cancer Inst. 2021;113(2):182-191.
doi pubmed pmc - Lees JG, White D, Keating BA, Barkl-Luke ME, Makker PGS, Goldstein D, Moalem-Taylor G. Oxaliplatin-induced haematological toxicity and splenomegaly in mice. PLoS One. 2020;15(9):e0238164.
doi pubmed pmc - Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, Nederlof I, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med. 2019;25(6):920-928.
doi pubmed - Scurr M, Pembroke T, Bloom A, Roberts D, Thomson A, Smart K, Bridgeman H, et al. Effect of modified vaccinia Ankara-5T4 and low-dose cyclophosphamide on antitumor immunity in metastatic colorectal cancer: a randomized clinical trial. JAMA Oncol. 2017;3(10):e172579.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.