

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Review
Volume 15, Number 3, June 2024, pages 372-381

Comprehensive Insights Into Renal Perivascular Epithelioid Cell Neoplasms: From Molecular Mechanisms to Clinical Practice
Bao Nan Donga ![]() , Hui Zhana, b, Ting Luana, Jian Song Wanga
, Hui Zhana, b, Ting Luana, Jian Song Wanga
aUrology Surgery Department, The Second Affiliated Hospital of Kunming Medical University, Kunming, Yunnan, China
bCorresponding Author: Hui Zhan, Urology Surgery Department, The Second Affiliated Hospital of Kunming Medical University, Kunming 650101, Yunnan, China
Manuscript submitted December 15, 2023, accepted March 16, 2024, published online April 15, 2024
Short title: Renal Perivascular Epithelioid Cell Neoplasms
doi: https://doi.org/10.14740/wjon1794
- Abstract
- Introduction
- Pathogenesis
- Pathological Features
- Clinical Manifestations and Diagnosis
- Therapeutic Options
- Clinical Case
- Conclusion
- References
| Abstract | ▴Top |
Perivascular epithelioid cell neoplasms (PEComas) are a rare category of mesenchymal tissue tumors, manifesting across various tissues and organs such as the kidneys, liver, lungs, pancreas, uterus, ovaries, and gastrointestinal tract. They predominantly affect females more than males. PEComas characteristically express both melanocytic and smooth muscle markers, making immunohistochemistry vital for their diagnosis. Renal angiomyolipoma (AML) represents a common variant of PEComas, typically marked by favorable prognoses. Nonetheless, only a small fraction of subtypes, especially epithelioid AML, possess the capacity to be malignant. Renal PEComas usually appear as asymptomatic masses accompanied by vague imaging characteristics. The main methods for diagnosis are histopathological analysis and the application of immunohistochemical stains. Presently, a uniform treatment plan for renal PEComas is absent. Strategies for management include active surveillance, selective arterial embolization, surgical procedures, and drug-based treatments. The focus of this review is on renal PEComas, shedding light on their pathogenesis, pathological characteristics, clinical presentations, diagnosis, and treatment modalities, and incorporating a clinical case study.
Keywords: Perivascular epithelioid cell neoplasms; Angiomyolipoma; Etiology; Immunohistochemistry; Therapeutics
| Introduction | ▴Top |
Perivascular epithelioid cell neoplasms (PEComas) are an uncommon type of tumor. According to the 2020 WHO Classification of Soft Tissues and Bone Tumors, PEComas are described as mesenchymal neoplasms composed of perivascular epithelioid cells (PECs) - distinctive epithelioid cells that are often closely associated with blood vessel walls and that express both melanocytic and smooth muscle markers [1]. PEComas can manifest in various body parts, including the kidneys, liver, lungs, pancreas, uterus, ovaries, and gastrointestinal tract, and exhibit a higher prevalence in women. Excluding organs linked to gender (like the uterus, uterine, prostate), the occurrence rate in women ranges from 1.6 to 5 times greater than in men, as reported by various institutions [1-4]. Diagnosis predominantly relies on histopathology, complemented by immunohistochemistry, which typically reveals melanocytic and smooth muscle markers. Currently, there is no standardized treatment protocol for PEComas, and the main therapeutic approach involves surgical resection and adjuvant drug therapy, with options like chemotherapy, tyrosine kinase inhibitors (TKIs), and the mechanistic target of rapamycin (mTOR) inhibitors.
This review comprehensively examines renal PEComas, including their pathogenesis, pathological characteristics, clinical presentations, diagnosis, and treatment modalities. The latter section offers a succinct overview of PEComas, framed within a recent case involving a male patient diagnosed with renal PEComas at our institution.
| Pathogenesis | ▴Top |
The precise mechanisms underlying the pathogenesis of PEComas remain elusive. Considering molecular genetics, the emergence of this tumor may be associated with mutations in specific genes including tuberous sclerosis complex (TSC), TFE3, and TP53.
TSC mutation
The TSC, an autosomal dominant disorder, arises from mutations in the TSC1/TSC2 genes, impacting multiple human organs and tissues including the brain, skin, heart, lungs, and kidneys. Its primary manifestations are neurological and psychiatric symptoms. Chromosome 9q34.13 houses TSC1, which encodes the hamartin protein, while chromosome 16p13.3 houses TSC2, which encodes the tuberin protein. The coding products of the two genes collaborate as a heterodimer in the creation of the TSC, classified as a tumor suppressor, thereby impeding the mTOR pathway. The normally active mTOR pathway facilitates cell growth and proliferation, and reduces autophagic cell death by engaging several downstream signaling molecules such as 4EBP1, S6K, SREBP1, and ULK1. Mutations in TSC1/TSC2 disrupt the TSC, leading to unchecked cell proliferation due to deregulation of the mTOR pathway [5, 6]. Following the exclusion of about 10% of TSC patients with mutations not detectable [6], close to 70% show TSC2 mutations and 20% exhibit TSC1 mutations [7, 8]. Additionally, chromosomal analysis of PEComas tumor tissues indicates frequent TSC2 gene deletions on chromosome 16p [9, 10], implicating mTOR pathway activation due to TSC gene mutations in PEComas pathogenesis. While there is a close association between TSC and PEComas, they are distinct conditions. Roughly 60% to 80% of TSC patients develop PEComas, while approximately 80% to 90% of PEComas patients do not concurrently present with TSC [11-13].
Microphthalmia-associated transcription factor (MiT) family
The MiT family, including MiT, TFEB, TFEC, and TFE3, plays a pivotal role in tumorigenesis by regulating autophagy and lysosomal functions. Amplifications and rearrangements of MiT, TFEB, and TFE3 within this family have been identified in various human tumors, including melanoma, renal cell carcinoma (RCC), and lung soft tissue sarcoma, among others [14, 15]. TFE3, located on chromosome Xp11, is frequently involved in gene fusion events due to chromosomal translocations, thus contributing to disease pathogenesis. In PEComas, TFE3 has been found to fuse with multiple genes, such as SFPQ, DVL2, NONO and RBMX [16-19]. Historically, the absence of concurrent TSC1/TSC2 and TFE3 alterations in PEComas led researchers to view TFE3 rearrangement as an alternative to TSC mutations, deemed mutually exclusive [20]. However, recent studies reveal the coexistence of TSC mutations and TFE3 overexpression in PEComas [21]. Another member of this familial cohort, MiT, has also demonstrated expression within PEComas. Pertinent investigations suggest that the overexpression of MiT could potentially stimulate the proliferation, invasion, and metastasis of PEComas by elevating the downstream expression levels of CYR61 [22].
TP53 mutation
A range of human cancers, PEComas included, show alterations in the TP53 oncogene [23]. Research into most PEComas instances has revealed that mutations in TP53 often occur alongside alterations in TSC1/TSC2, TFE3, and other genes involved in PEComa development, with these gene mutations not being the sole cause of PEComas [24-26]. Recent research has pinpointed PEComas with TP53 mutations, absent of simultaneous TSC mutations or TFE3 rearrangements [27], indicating TP53’s possible role as a key influencer in PEComa’s emergence. Nonetheless, additional studies are required to determine if mutations in TP53 solely serve as the fundamental cause of PEComas without TSC mutations or TFE3 rearrangements [28, 29].
To sum up, the pathogenic mechanisms underlying PEComas remain elusive. Current analysis of case studies indicates that PEComas’ etiology is not attributable to a singular gene mutation. Instead, it appears to result from a confluence of multiple genetic alterations, notably involving TSC1/TSC2, TFE3, and TP53 genes. Further investigative efforts are imperative to elucidate the precise pathogenesis of PEComas.
| Pathological Features | ▴Top |
PEComas tissue characteristically contains a plethora of blood vessels and a significant number of epithelioid cells (PECs). Due to the absence of an analogous cell in normal human tissues, the origin of PEComas is still indeterminate, with some experts suggesting a neural crest origin. PECs are typically arranged in radial or clustered patterns around blood vessels, frequently penetrating into the walls of small to medium-sized vessels, extending to the subendothelial layer. PECs adjacent to vessel walls predominantly exhibit an epithelioid form, whereas those more distant present as spindle-shaped. The PEC cytoplasm is eosinophilic, turning translucent when it accumulates substantial fat. The cells’ central nuclei are diminutive, round, or oval, sometimes encircled by an eosinophilic band. Nucleoli are minute and pronounced, and certain PECs may display intensely stained nucleoli or irregular nuclear morphology [2, 30]. Renal angiomyolipoma (AML) is a common type of PEComas. AML’s cancerous tissue contains typical PECs and is rich in vascular and adipose components, accompanied by irregularly distributed epithelioid or spindle-shaped smooth muscle cells. Renal AML is categorized into various types such as classic AML, microscopic AMLs, intraglomerular lesions with similar features of AML, AML with epithelial cysts, oncocytoma-like AMLs, lymphangiomyomatosis of the renal sinus, epithelioid AML (eAML), among others, due to differences in composition, tissue structure, and lesion placement [13, 31]. Most renal PEComas are non-malignant growths, typically showing positive outcomes. Merely a select few exhibit a prognosis that is more pessimistic. AMLs prone to malignancy often exhibit harmful pathological traits, signaling invasive tendencies, such as pronounced nuclear atypia, elevated cell density, a high nuclear division index, tumor necrosis, and the invasion of blood vessels and lymphatic systems [2, 13, 30, 32]. eAML, a variant of AML, refers to a classification with malignant potential [32, 33-34], accounting for approximately 4.6% of all AML instances. Epithelioid cell percentages in eAML vary between 5% and 100%, which may correlate with the severity of the tumor’s malignancy [31, 35]. Various research indicates specific baseline ratios of epithelioid cells necessary for eAML diagnosis. According to the 2022 WHO tumor classification, it is advised to diagnose eAML if the proportion of epithelioid cells is 80% or more [36].
The prognosis of various eAML cases varies, discernible through their histopathological features, indicating diverse malignancy or risk groups. Brimo et al identified four adverse characteristics: 1) ≥ 70% atypical epithelioid cells; 2) ≥ 2 mitotic figures per 10 high-power fields (HPF); 3) atypical mitotic figures; and 4) necrosis. eAML presenting with 1 - 2 of these adverse features is classified as benign, while those manifesting 3 - 4 features are considered malignant [4]; Nese et al delineated five criteria: 1) TSC and/or concurrent AML; 2) tumor size (> 7 cm); 3) morphological pattern A; 4) extrarenal extension and/or involvement of renal vein; and 5) necrosis. Meeting 0 - 1 of these criteria signifies a low-risk group, 2 - 3 criteria indicate a moderate-risk group, and meeting 4 - 5 criteria corresponds to a high-risk group [37].
Immunohistochemistry indicates that most PEComas simultaneously express markers of melanocytic markers (such as HMB45, melan-A, MiT) and smooth muscle markers (such as SMA, desmin, caldesmon, etc.). Other commonly expressed markers include cathepsin K [38, 39], STING [40], PNL2 [41], TFE3, S100, etc. [13, 31]. In some PEComas, only one type of marker may be expressed or predominance in the expression of one over the other is observed, such as epithelioid cell-dominant PEComas tending to highly express melanocytic markers, while spindle cell-dominant PEComas exhibit high expression of smooth muscle markers [2]. In renal PEComas, markers with higher sensitivity include HMB45 and melan-A [42, 43]. Other common indicators include estrogen receptor (ER) and progesterone receptor (PR) [13], with literature indicating that ER and PR positivity rates fluctuate between roughly 42.4% and 83% and 15.2% and 100%, respectively [44, 45]. Other commonly noted markers include PNL2, cathepsin K, and more. eAML’s marker profile, indicative of malignancy, reflects that of other AMLs [41, 43], characterized by markers such as Ki-67 [46], p53 [26], SMA [47], which show varied expression levels and could be significant in prognosis.
| Clinical Manifestations and Diagnosis | ▴Top |
Renal PEComas usually appear asymptomatically and are frequently found by chance in imaging processes [11]. TSC-related AML typically begins at a younger age, as various studies show a median age under 20 years [12, 48, 49], with about 80% of such cases presenting as bilateral, multifocal masses, and tumors not exceeding 3 cm in diameter accounting for roughly 65% of these cases. The distinct manifestation of small, multifocal, bilateral masses becomes more evident in those with TSC2 mutations [12, 48]. Excluding approximately 80-90% of patients without kidney symptoms, common indicators of TSC-related AML include nonspecific pain, high blood pressure, tumor rupture causing bleeding, blood in urine, and reduced kidney function. Cases of symptomatic AML usually appear in the younger population, mainly marked by TSC2 mutations [48, 49]. Sporadic AML typically begins around the age of 50, with unilateral solitary tumors being the predominant manifestation, and significant differences exist in the largest tumor sizes recorded among various medical centers [3, 50-52]. Roughly 50% patients with sporadic AML show symptoms, frequently reflecting the kidney-related symptoms observed in TSC-related AML [32], with a higher incidence of tumor rupture and hemorrhage in large-volume sporadic AML cases [50, 51]. Comparative research differentiating pathological subtypes indicates a notably larger average tumor size in eAML compared to non-eAML in the control group [3, 33, 50], suggesting an increased likelihood of tumor rupture and hemorrhage in large-volume eAML.
The imaging of renal PEComas lacks specificity [53]. Renal PEComas may exhibit hypointense or isointense shadows in non-enhanced computed tomography (CT), while in enhanced CT, PEComas demonstrate notable enhancement in the arterial and venous phases, and slight enhancement in the delayed phase. Conversely, in magnetic resonance imaging (MRI), renal PEComas display hypointense or isointense shadows in T1-weighted images, inhomogeneous hyperintense shadows in T2-weighted images, and substantial enhancement following enhanced scanning [52, 54]. Classic AML is characterized by abundant fatty tissue, blood vessels, and smooth muscle tissue. It demonstrates attenuation patterns resembling fatty tissue on CT scans. On frequency-selective fat suppression MRI and chemical shift MRI, it exhibits the loss of signals. Fat-poor AML, owing to its low fat content, presents imaging characteristics akin to RCC, posing challenges in differentiation. Several machine learning models utilizing non-enhanced CT texture features have shown promise in distinguishing fat-poor AML from RCC, achieving high accuracy with area under the curve (AUC) values exceeding 0.80 [55, 56]. eAML may present as high-density shadows on CT scans, often accompanied by irregular enhancement or cystic formations, and appears as low signal intensity on T2-weighted MRI [53, 54, 57, 58]. Diagnosing PEComas solely through CT and MRI is a challenging task, and research has indicated that the accuracy of CT and MRI in detecting PEComas prior to surgery ranges from 15% to 31% and 22% to 40%, respectively [52, 59]. Although imaging plays a limited role in diagnosing and treating PEComas, it is important not to overlook its two functions: firstly, it can aid in the initial assessment of tumor benignity or malignancy by measuring the size of the primary tumor and detecting necrosis, hemorrhage, invasion of surrounding tissues or blood vessels, enlarged lymph nodes, and other malignant tendencies of the tumor through CT and MRI; secondly, it can identify recurrent metastatic foci during post-treatment follow-up, such as the utilization of positron emission tomography (PET)-CT to detect the elevated concentration of 18F-FDG in metastatic foci of PEComas [53, 60].
| Therapeutic Options | ▴Top |
Given the generally low malignancy risk associated with most renal PEComas subtypes, active surveillance (AS) emerges as a predominant treatment approach [61]. A study involving 130 AML patients uncovered that 13% of those initially opting for AS transitioned to active treatment after an average monitoring duration of 49 months. The COX regression analysis highlighted that tumors exceeding 4 cm in size and the onset of symptoms related to the tumor played pivotal roles in the decision to discontinue AS [62]. A subsequent meta-analysis indicated that during the follow-up period, 11% of AML patients under AS experienced an increase in tumor size, 2.2% suffered from spontaneous bleeding or hematuria, and 5.7% underwent active intervention. This intervention was primarily carried out through selective arterial embolization (77%), followed by surgery (19%), and ultimately radiofrequency ablation (4%) [63]. Selective arterial embolization represents a secure and minimally invasive treatment approach; however, there is a recurrence rate ranging from 25% to 40% following arterial embolization for AML. This recurrence rate escalates notably when dealing with the embolization of large-volume renal AMLs exceeding 8 cm in diameter [64, 65]. Surgical resection of tumor lesions emerges as another effective treatment modality, with the specific surgical plan contingent on factors such as the patient’s baseline condition, tumor location, and size. Preferably, partial nephrectomy should be prioritized to preserve renal units. In the case of renal PEComas, considering drug therapy is also a viable option.
A wide range of chemotherapeutic drugs, including anthracycline, platinum, gemcitabine, etc., are accessible for renal PEComas. In light of the abundant vascularity of renal PEComas, certain scholars have opted to employ anti-vascular endothelial growth factor TKIs, such as apatinib [66], sunitinib [67], imatinib [68], etc., for the management of PEComas. Given the potential correlation between the pathogenesis of PEComas and mTOR, the utilization of mTOR inhibitors (e.g. everolimus, sirolimus, temsirolimus, etc.) has yielded positive outcomes as well [69]. Zonnenberg et al revealed that following a 2-year regimen of everolimus, a decrease in kidney size was observed in 85.2% of patients (compared to 37.9% in the control group, P = 0.0003), and the mean kidney size in the treatment group diminished by 8.8 mm (as opposed to 1.7 mm in the control group, P = 0.01). The mean time to best response in kidney size was 8.2 months in the treatment group (vs. 14.1 months in the control group, P = 0.0003) [70]. The study conducted by Cai et al demonstrates that the median response time for treating TSC-related AML with everolimus is 3 months. Following a year of therapy, the tumor size diminished to 41% of its initial volume (P < 0.002). Yet, after stopping the treatment, the tumor size rose to 67% (P = 0.006) and 78% (P = 0.014) of its initial size at 6 and 12 months, in that order. The results indicate the need for prolonged use of everolimus treatment in treating TSC-related AML [71]. Another study using sirolimus to treat both TSC-related and sporadic AML noted a reduction in the median tumor size of AML to half of its initial levels post-intervention. Significantly, the decrease was more marked in tumors with low fat content, showing a reduction of -67%, in contrast to a -15% decrease in tumors with high fat content (P < 0.001) [72]. Sanfilippo et al [73] conducted a statistical analysis on the impact of various medications on 53 patients with progressive PEComas. The findings indicate that anthracyclines exhibit a 13% objective remission rate (ORR), a median progression-free survival (mPFS) of 3.2 months, gemcitabine demonstrates a 20% ORR and a mPFS of 3.4 months for PEComa, whereas TKIs display an ORR and mPFS of 8.3% and 5.4 months, respectively. The mTOR inhibitors (everolimus 12.5%, sirolimus 80%, and temsirolimus 7.5%) exhibited the highest efficacy with an ORR of 41% and a mPFS of 9 months. Due to the absence of simultaneous occurrence of TSC1/TSC2 mutations and TFE3 rearrangements in the majority of PEComas, certain studies have proposed that TFE3 rearrangements and TSC1/TSC2 mutations are mutually exclusive, thereby suggesting that PEComa with TFE3 rearrangement exhibits insensitivity towards mTOR inhibitors [20]. Hence, when diagnosing and treating PEComa, it is crucial to meticulously choose the drug treatment plan based on its potential combination with TFE3 rearrangement. To sum up, a variety of treatments exist for renal PEComas, and medical professionals ought to take into account the unique circumstances of each patient prior to deciding on renal PEComa treatments.
| Clinical Case | ▴Top |
A 27-year-old male patient was referred to our hospital for evaluation of a right kidney mass, initially detected during a routine health checkup at a local hospital. The patient reported no symptoms such as abdominal or back pain, hematuria, urinary frequency or urgency, dizziness, palpitations, fatigue, or poor appetite. Physical examination revealed a flat and soft abdomen, with no abnormal pressure points, palpable masses or nodules, vertebrocostal point or lumbocostal point tenderness, and no renal region tenderness.
CT imaging of both kidneys displayed multiple nodular masses of medium and slightly high density, the largest located in the upper pole of the right kidney, measuring approximately 7.9 × 7.6 cm. The mass displaced surrounding tissues but maintained clear boundaries. The enhancement scan showed inhomogeneous enhancement of the mass, with an increased number of right renal artery branches and multiple small cystic shadows in the right renal pole. MRI revealed a mass at the upper pole of the right kidney, characterized by high T2-weighted image and mixed T1-weighted image signals. Diffusion-weighted imaging (DWI) and apparent diffusion coefficient (ADC) indicated mixed high and low signals. Mild, uneven enhancement was observed during both the parenchymal and excretory phases, with some boundaries being indistinct and compressing adjacent tissues. The small nodules in both kidneys exhibited high T2 and slightly elevated T1 signals, showing uneven enhancement following contrast administration. These findings raise the possibility of RCC or lipid-poor AML at the right kidney’s upper pole (Fig. 1). Routine blood tests, liver and kidney function assessments, electrolyte levels, coagulation profiles, renin-angiotensin system evaluations, and adrenocorticotropic hormone measurements were all within normal limits. The estimated glomerular filtration rate (eGFR) registered at 123 mL/min.
 Click for large image | Figure 1. The computed tomography (CT) and magnetic resonance imaging (MRI) of the patient. |
The left renal nodule was initially addressed with an ultrasound-guided puncture biopsy and radiofrequency ablation. Pathological examination indicated that it was fibro-fatty tissue without any epithelial component or malignant features. Immunohistochemistry results showed VIM (-), PAX-8 (-), CD68 (-), CD163 (-), CK (-), HMB45 (-), melan-A (-), and Ki67 (< 1%). Seven days later, a transabdominal laparoscopic radical nephrectomy of the right kidney was performed. During the surgery, a large mass with a rich blood supply was observed at the upper pole of the right kidney. It was poorly demarcated from the normal renal tissues and had formed severe adhesions with the lower lobe of the liver, the descending part of the duodenum, and the inferior vena cava. The mass was meticulously dissected, completely separated, and then excised along with the right kidney (Fig. 2).
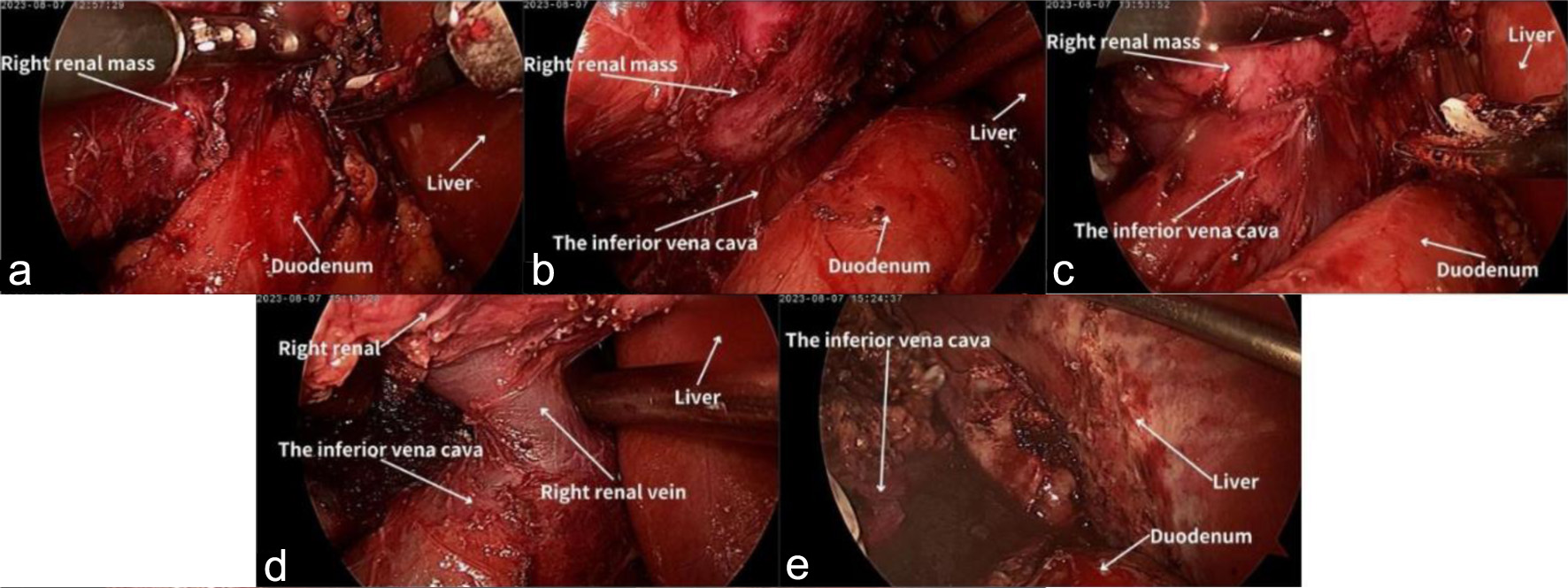 Click for large image | Figure 2. The radical nephrectomy of the right kidney. (a) Adhesion of the right kidney to the descending portion of the duodenum. (b) The kidney and the duodenum after adhesion were released. (c) Adhesion of the right kidney to the inferior vena cava. (d) The kidney and the inferior vena cava after adhesion were released. (e) The lower lobe of the liver after adhesion was released. |
Postoperative pathological examination revealed that the right renal mass was PEComa. This diagnosis was characterized by a predominance of atypical epithelioid cells, significant nuclear atypia exceeding 2/10 HPF, pathological mitotic figures, focal tumor necrosis, and vascular invasion. Immunohistochemical analysis showed positive staining for HMB45, melan-A, and PNL2, but negative for desmin, S100, and TFE3, further supporting the PEComas diagnosis (Fig. 3). The final diagnosis of malignant PEComa in the right kidney was established based on these histological and immunohistochemical findings. Postoperatively, the patient recovered well without major complications such as severe bleeding, infection, renal insufficiency, or urinary leakage. A flowchart of the patient’s main treatment events is shown in Figure 4. The patient presented solely with a one-sided renal mass identified as PEComas, while the opposite renal mass was not renal PEComas, and no additional irregularities were found in the physical exam or supplementary examination, failing to align with the clinical diagnostic criteria of TSC [74]. Post-surgery, our recommendation was for the patient to try a genetic screening for TSC-related mutations, yet the patient declined any additional genetic testing for personal reasons. The instructions were given for the patient to consume everolimus by oral, and the subsequent check-up period has spanned roughly 7 months, currently free from any clear adverse effects, and the tumor did not recur metastatically in the chest and abdominal CT scans.
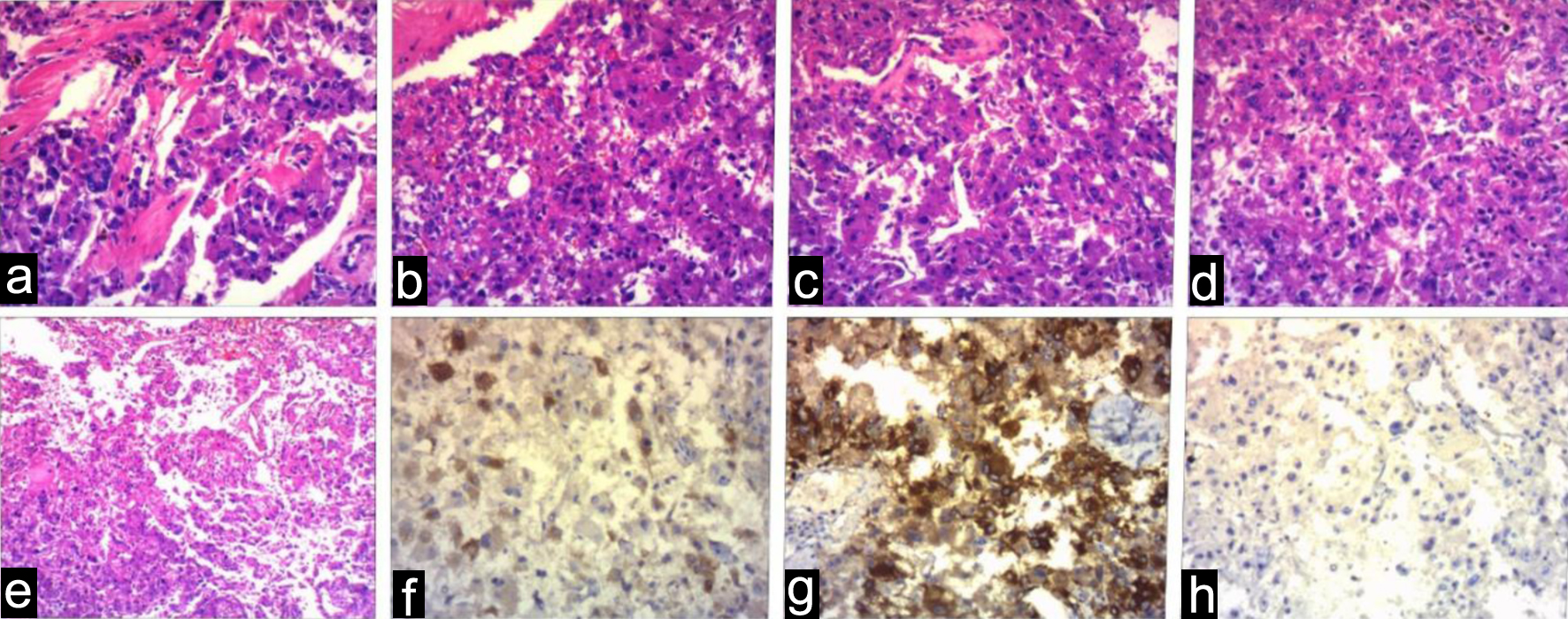 Click for large image | Figure 3. The histopathology (a-e) and immunohistochemistry (f-h) of the right renal mass. (f) HMB45 positive, (g) melan-A positive, (h) TFE3 negative. |
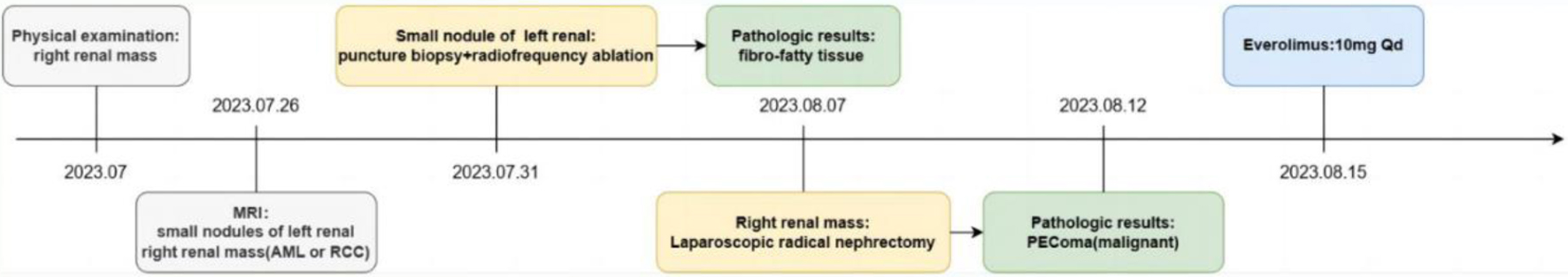 Click for large image | Figure 4. The case history. |
| Conclusion | ▴Top |
Renal PEComas represent a rare tumor variety, marked by the lack of distinctive imaging characteristics. Consequently, the primary method for their diagnosis is based on histopathological and immunohistochemical techniques. Typically, PEComas express both melanocytic and smooth muscle markers. Most renal PEComas correlate with positive results, hence the preference for active surveillance as a treatment option. When patients show tumor-related symptoms and their tumors grow larger, active treatment becomes feasible, including selective arterial embolization, surgical procedures, radiofrequency ablation, among others. Treatment with medication can also be used for renal PEComas management. Currently, the efficacy of mTOR inhibitors, such as everolimus and sirolimus, in treating renal PEComas is more pronounced.
Acknowledgments
None to declare.
Financial Disclosure
This work was supported by the National Natural Science Foundation of China (No. 82060464) and the Yunnan Science and Technology Department and Kunming Medical University Special Fund (grant No. 202001AY070001-148).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Informed Consent
Written informed consent was obtained from the patient, all patient information was deidentified.
Author Contributions
Bao Nan Dong (M.M.): author, data collection, formal analysis, writing-original draft. Hui Zhan (M.D.): operator, conceptualization, resources, data curation. Ting Luan (M.D.): writing-review and editing, funding acquisition. Jian Song Wang (M.D.): supervision, funding acquisition. All authors read and approved the final version.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- The WHO Classification of Tumours Editorial Board. WHO Classification of Tumours Soft Tissue and Bone Tumours, 5th Ed. Lyon: IARC Press; 2020.
- Folpe AL, Mentzel T, Lehr HA, Fisher C, Balzer BL, Weiss SW. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol. 2005;29(12):1558-1575.
doi pubmed - Kaneko K, Yoshida S, Yamamoto K, Arita Y, Kijima T, Yokoyama M, Ishioka J, et al. Renal epithelioid angiomyolipoma: Incidence in a Japanese cohort and diagnostic utility of diffusion-weighted magnetic resonance imaging. Int J Urol. 2020;27(7):599-604.
doi pubmed - Brimo F, Robinson B, Guo C, Zhou M, Latour M, Epstein JI. Renal epithelioid angiomyolipoma with atypia: a series of 40 cases with emphasis on clinicopathologic prognostic indicators of malignancy. Am J Surg Pathol. 2010;34(5):715-722.
doi pubmed - Salussolia CL, Klonowska K, Kwiatkowski DJ, Sahin M. Genetic etiologies, diagnosis, and treatment of tuberous sclerosis complex. Annu Rev Genomics Hum Genet. 2019;20:217-240.
doi pubmed - Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035.
doi pubmed - Dabora SL, Jozwiak S, Franz DN, Roberts PS, Nieto A, Chung J, Choy YS, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68(1):64-80.
doi pubmed pmc - Lam HC, Siroky BJ, Henske EP. Renal disease in tuberous sclerosis complex: pathogenesis and therapy. Nat Rev Nephrol. 2018;14(11):704-716.
doi pubmed - Pan CC, Jong YJ, Chai CY, Huang SH, Chen YJ. Comparative genomic hybridization study of perivascular epithelioid cell tumor: molecular genetic evidence of perivascular epithelioid cell tumor as a distinctive neoplasm. Hum Pathol. 2006;37(5):606-612.
doi pubmed - Pan CC, Chung MY, Ng KF, Liu CY, Wang JS, Chai CY, Huang SH, et al. Constant allelic alteration on chromosome 16p (TSC2 gene) in perivascular epithelioid cell tumour (PEComa): genetic evidence for the relationship of PEComa with angiomyolipoma. J Pathol. 2008;214(3):387-393.
doi pubmed - Ritter DM, Fessler BK, Ebrahimi-Fakhari D, Wei J, Franz DN, Krueger DA, Trout AT, et al. Prevalence of thoracoabdominal imaging findings in tuberous sclerosis complex. Orphanet J Rare Dis. 2022;17(1):124.
doi pubmed pmc - Rakowski SK, Winterkorn EB, Paul E, Steele DJ, Halpern EF, Thiele EA. Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors. Kidney Int. 2006;70(10):1777-1782.
doi pubmed - Thway K, Fisher C. PEComa: morphology and genetics of a complex tumor family. Ann Diagn Pathol. 2015;19(5):359-368.
doi pubmed - Yang M, Liu E, Tang L, Lei Y, Sun X, Hu J, Dong H, et al. Emerging roles and regulation of MiT/TFE transcriptional factors. Cell Commun Signal. 2018;16(1):31.
doi pubmed pmc - Ploper D, De Robertis EM. The MITF family of transcription factors: Role in endolysosomal biogenesis, Wnt signaling, and oncogenesis. Pharmacol Res. 2015;99:36-43.
doi pubmed - Shon W, Kim J, Sukov W, Reith J. Malignant TFE3-rearranged perivascular epithelioid cell neoplasm (PEComa) presenting as a subcutaneous mass. Br J Dermatol. 2016;174(3):617-620.
doi pubmed - McGregor SM, Alikhan MB, John RA, Kotler H, Bridge JA, Mujacic I, Kadri S, et al. Melanotic PEComa of the sinonasal mucosa with NONO-TFE3 fusion: an elusive mimic of sinonasal melanoma. Am J Surg Pathol. 2017;41(5):717-722.
doi pubmed - Shen Q, Rao Q, Xia QY, Yu B, Shi QL, Zhang RS, Zhou XJ. Perivascular epithelioid cell tumor (PEComa) with TFE3 gene rearrangement: clinicopathological, immunohistochemical, and molecular features. Virchows Arch. 2014;465(5):607-613.
doi pubmed - Argani P, Zhong M, Reuter VE, Fallon JT, Epstein JI, Netto GJ, Antonescu CR. TFE3-fusion variant analysis defines specific clinicopathologic associations among Xp11 translocation cancers. Am J Surg Pathol. 2016;40(6):723-737.
doi pubmed pmc - Agaram NP, Sung YS, Zhang L, Chen CL, Chen HW, Singer S, Dickson MA, et al. Dichotomy of genetic abnormalities in PEComas with therapeutic implications. Am J Surg Pathol. 2015;39(6):813-825.
doi pubmed pmc - Schmiester M, Dolnik A, Kornak U, Pfitzner B, Hummel M, Treue D, Hartmann A, et al. TFE3 activation in a TSC1-altered malignant PEComa: challenging the dichotomy of the underlying pathogenic mechanisms. J Pathol Clin Res. 2021;7(1):3-9.
doi pubmed pmc - Zarei M, Giannikou K, Du H, Liu HJ, Duarte M, Johnson S, Nassar AH, et al. MITF is a driver oncogene and potential therapeutic target in kidney angiomyolipoma tumors through transcriptional regulation of CYR61. Oncogene. 2021;40(1):112-126.
doi pubmed pmc - Wang H, Guo M, Wei H, Chen Y. Targeting p53 pathways: mechanisms, structures, and advances in therapy. Signal Transduct Target Ther. 2023;8(1):92.
doi pubmed pmc - Selenica P, Conlon N, Gonzalez C, Frosina D, Jungbluth AA, Beets-Tan RGH, Rao MK, et al. Genomic profiling aids classification of diagnostically challenging uterine mesenchymal tumors with myomelanocytic differentiation. Am J Surg Pathol. 2021;45(1):77-92.
doi pubmed pmc - Agaram NP, Sung YS, Zhang L, Chen CL, Chen HW, Berger M, Antonescu C. Memorial Sloan Kettering Cancer Center, New York, NY, United States. Genomic characterization of pecomas: Dichotomy of genetic abnormalities with therapeutic implications. Lab Invest. 2015;95:12A-13A.
doi - Zhang Y, Wei X, Teng X, Chen G. p53 aberration and TFE3 gene amplification may be predictors of adverse prognosis in epithelioid angiomyolipoma of the kidney. Diagn Pathol. 2023;18(1):14.
doi pubmed pmc - Butz H, Lovey J, Szentkereszty M, Bozsik A, Toth E, Patocs A. Case report: a novel pathomechanism in PEComa by the loss of heterozygosity of TP53. Front Oncol. 2022;12:849004.
doi pubmed pmc - Boudaouara O, Kallel R, Dhieb D, Smaoui W, Ayed HB, Keskes L, Sellami Boudawara T. Renal angiomyolipoma: Clinico-pathologic study of 17 cases with emphasis on the epithelioid histology and p53 gene abnormalities. Ann Diagn Pathol. 2020;47:151538.
doi pubmed - Cho JH, Patel B, Bonala S, Mansouri H, Manne S, Vadrevu SK, Ghouse S, et al. The codon 72 TP53 polymorphism contributes to TSC tumorigenesis through the notch-nodal axis. Mol Cancer Res. 2019;17(8):1639-1651.
doi pubmed pmc - Folpe AL, Kwiatkowski DJ. Perivascular epithelioid cell neoplasms: pathology and pathogenesis. Hum Pathol. 2010;41(1):1-15.
doi pubmed - Martignoni G, Pea M, Zampini C, Brunelli M, Segala D, Zamboni G, Bonetti F. PEComas of the kidney and of the genitourinary tract. Semin Diagn Pathol. 2015;32(2):140-159.
doi pubmed - Tsai HY, Lee KH, Ng KF, Kao YT, Chuang CK. Clinicopathologic analysis of renal epithelioid angiomyolipoma: Consecutively excised 23 cases. Kaohsiung J Med Sci. 2019;35(1):33-38.
doi pubmed - Lee W, Choi SY, Lee C, Yoo S, You D, Jeong IG, Song C, et al. Does epithelioid angiomyolipoma have poorer prognosis, compared with classic angiomyolipoma? Investig Clin Urol. 2018;59(6):357-362.
doi pubmed pmc - Park JH, Lee C, Suh JH, Kim G, Song B, Moon KC. Renal epithelioid angiomyolipoma: Histopathologic review, immunohistochemical evaluation and prognostic significance. Pathol Int. 2016;66(10):571-577.
doi pubmed - Lei JH, Liu LR, Wei Q, Song TR, Yang L, Yuan HC, Jiang Y, et al. A four-year follow-up study of renal epithelioid angiomyolipoma: a multi-center experience and literature review. Sci Rep. 2015;5:10030.
doi pubmed pmc - Board WC of TE. Urinary and Male Genital Tumours. Accessed March 12, 2024. https://publications.iarc.fr/Book-And-Report-Series/Who-Classification-Of-Tumours/Urinary-And-Male-Genital-Tumours-2022.
- Nese N, Martignoni G, Fletcher CD, Gupta R, Pan CC, Kim H, Ro JY, et al. Pure epithelioid PEComas (so-called epithelioid angiomyolipoma) of the kidney: A clinicopathologic study of 41 cases: detailed assessment of morphology and risk stratification. Am J Surg Pathol. 2011;35(2):161-176.
doi pubmed - Martignoni G, Bonetti F, Chilosi M, Brunelli M, Segala D, Amin MB, Argani P, et al. Cathepsin K expression in the spectrum of perivascular epithelioid cell (PEC) lesions of the kidney. Mod Pathol. 2012;25(1):100-111.
doi pubmed - Rao Q, Cheng L, Xia QY, Liu B, Li L, Shi QL, Shi SS, et al. Cathepsin K expression in a wide spectrum of perivascular epithelioid cell neoplasms (PEComas): a clinicopathological study emphasizing extrarenal PEComas. Histopathology. 2013;62(4):642-650.
doi pubmed - Calio A, Brunelli M, Gobbo S, Pedron S, Segala D, Argani P, Martignoni G. Stimulator of interferon genes (STING) immunohistochemical expression in the spectrum of perivascular epithelioid cell (PEC) lesions of the kidney. Pathology. 2021;53(5):579-585.
doi pubmed - Gulavita P, Fletcher CDM, Hirsch MS. PNL2: an adjunctive biomarker for renal angiomyolipomas and perivascular epithelioid cell tumours. Histopathology. 2018;72(3):441-448.
doi pubmed - Hohensee SE, La Rosa FG, Homer P, Suby-Long T, Wilson S, Lucia SM, Iczkowski KA. Renal epithelioid angiomyolipoma with a negative premelanosome marker immunoprofile: a case report and review of the literature. J Med Case Rep. 2013;7:118.
doi pubmed pmc - Fejes Z, Santa F, Jenei A, Kiraly IE, Varga L, Kuthi L. Angiomyolipoma of the kidney-Clinicopathological analysis of 52 cases. Pathol Oncol Res. 2022;28:1610831.
doi pubmed pmc - Cho NH, Shim HS, Choi YD, Kim DS. Estrogen receptor is significantly associated with the epithelioid variants of renal angiomyolipoma: a clinicopathological and immunohistochemical study of 67 cases. Pathol Int. 2004;54(7):510-515.
doi pubmed - Logginidou H, Ao X, Russo I, Henske EP. Frequent estrogen and progesterone receptor immunoreactivity in renal angiomyolipomas from women with pulmonary lymphangioleiomyomatosis. Chest. 2000;117(1):25-30.
doi pubmed - Xu C, Jiang XZ, Zhao HF, Zhang NZ, Ma L, Xu ZS. The applicability of Ki-67 marker for renal epithelioid angiomyolipoma: experience of ten cases from a single center. Neoplasma. 2013;60(2):209-214.
doi pubmed - Anwaier A, Xu WH, Tian X, Ding T, Su JQ, Wang Y, Qu YY, et al. club suit symbolEvaluation of clinicopathological profiles and development of a risk model in renal epithelioid angiomyolipoma patients: a large-scale retrospective cohort study. BMC Urol. 2022;22(1):148.
doi pubmed pmc - Kingswood JC, Belousova E, Benedik MP, Carter T, Cottin V, Curatolo P, Dahlin M, et al. Renal angiomyolipoma in patients with tuberous sclerosis complex: findings from the TuberOus SClerosis registry to increase disease Awareness. Nephrol Dial Transplant. 2019;34(3):502-508.
doi pubmed pmc - Janssens P, Van Hoeve K, De Waele L, De Rechter S, Claes KJ, Van de Perre E, Wissing KM, et al. Renal progression factors in young patients with tuberous sclerosis complex: a retrospective cohort study. Pediatr Nephrol. 2018;33(11):2085-2093.
doi pubmed - Lee KH, Tsai HY, Kao YT, Lin HC, Chou YC, Su SH, Chuang CK. Clinical behavior and management of three types of renal angiomyolipomas. J Formos Med Assoc. 2019;118(1 Pt 1):162-169.
doi pubmed - Ruud Bosch JLH, Vekeman F, Duh MS, Neary M, Magestro M, Fortier J, Karner P, et al. Factors associated with the number and size of renal angiomyolipomas in sporadic angiomyolipoma (sAML): a study of adult patients with sAML managed in a Dutch tertiary referral center. Int Urol Nephrol. 2018;50(3):459-467.
doi pubmed pmc - Tan Y, Zhang H, Xiao EH. Perivascular epithelioid cell tumour: dynamic CT, MRI and clinicopathological characteristics—analysis of 32 cases and review of the literature. Clin Radiol. 2013;68(6):555-561.
doi pubmed - Phillips CH, Keraliya AR, Shinagare AB, Ramaiya NH, Tirumani SH. Update on the imaging of malignant perivascular epithelioid cell tumors (PEComas). Abdom Radiol (NY). 2016;41(2):368-376.
doi pubmed - Tirumani SH, Shinagare AB, Hargreaves J, Jagannathan JP, Hornick JL, Wagner AJ, Ramaiya NH. Imaging features of primary and metastatic malignant perivascular epithelioid cell tumors. AJR Am J Roentgenol. 2014;202(2):252-258.
doi pubmed - Feng Z, Rong P, Cao P, Zhou Q, Zhu W, Yan Z, Liu Q, et al. Machine learning-based quantitative texture analysis of CT images of small renal masses: Differentiation of angiomyolipoma without visible fat from renal cell carcinoma. Eur Radiol. 2018;28(4):1625-1633.
doi pubmed - Yao H, Tian L, Liu X, Li S, Chen Y, Cao J, Zhang Z, et al. Development and external validation of the multichannel deep learning model based on unenhanced CT for differentiating fat-poor angiomyolipoma from renal cell carcinoma: a two-center retrospective study. J Cancer Res Clin Oncol. 2023;149(17):15827-15838.
doi pubmed pmc - Jinzaki M, Silverman SG, Akita H, Mikami S, Oya M. Diagnosis of renal angiomyolipomas: classic, fat-poor, and epithelioid types. Semin Ultrasound CT MR. 2017;38(1):37-46.
doi pubmed - Wang C, Li X, Peng L, Gou X, Fan J. An update on recent developments in rupture of renal angiomyolipoma. Medicine (Baltimore). 2018;97(16):e0497.
doi pubmed pmc - Baez JC, Landry JM, Saltzman JR, Qian X, Zinner MJ, Mortele KJ. Pancreatic PEComa (sugar tumor): MDCT and EUS features. JOP. 2009;10(6):679-682.
pubmed - Sun L, Sun X, Li Y, Xing L. The role of (18)F-FDG PET/CT imaging in patient with malignant PEComa treated with mTOR inhibitor. Onco Targets Ther. 2015;8:1967-1970.
doi pubmed pmc - Nason GJ, Morris J, Bhatt JR, Richard PO, Martin L, Ajib K, Tan GH, et al. Natural history of renal angiomyolipoma favors surveillance as an initial approach. Eur Urol Focus. 2021;7(3):582-588.
doi pubmed - Ouzaid I, Autorino R, Fatica R, Herts BR, McLennan G, Remer EM, Haber GP. Active surveillance for renal angiomyolipoma: outcomes and factors predictive of delayed intervention. BJU Int. 2014;114(3):412-417.
doi pubmed - Fernandez-Pello S, Hora M, Kuusk T, Tahbaz R, Dabestani S, Abu-Ghanem Y, Albiges L, et al. Management of sporadic renal angiomyolipomas: a systematic review of available evidence to guide recommendations from the European Association of Urology Renal Cell Carcinoma Guidelines Panel. Eur Urol Oncol. 2020;3(1):57-72.
doi pubmed - Sward J, Bohlin K, Henrikson O, Lundstam S, Peeker R, Grenabo Bergdahl A. Long-term efficacy of selective arterial embolisation of renal angiomyolipoma. Scand J Urol. 2023;58:86-92.
doi pubmed - Kuusk T, Biancari F, Lane B, Tobert C, Campbell S, Rimon U, D'Andrea V, et al. Treatment of renal angiomyolipoma: pooled analysis of individual patient data. BMC Urol. 2015;15:123.
doi pubmed pmc - Zhang N, Ren Y, Zan L, Zhang X, Zhao J, Wen L, Wang Y. Case report: Kidney perivascular epithelioid cell tumor treated with anti-VEGFR tyrosine kinase inhibitor and MTOR inhibitor. Front Oncol. 2022;12:966818.
doi pubmed pmc - AlAzab RS, Alorjani MS, Sahawneh FE, Al-Sukhun S. Metastatic perivascular epithelioid cell tumor of the kidney: a case report with emphasis on response to the Tyrosine-Kinase inhibitor sunitinib. Res Rep Urol. 2019;11:311-317.
doi pubmed pmc - Rigby H, Yu W, Schmidt MH, Fernandez CV. Lack of response of a metastatic renal perivascular epithelial cell tumor (PEComa) to successive courses of DTIC based-therapy and imatinib mesylate. Pediatr Blood Cancer. 2005;45(2):202-206.
doi pubmed - Jia R, Jiang L, Zhou Y, Wang Y, Guo X, Ji Y, Ni X, et al. Clinical features of 18 perivascular epithelioid cell tumor cases. Medicine (Baltimore). 2020;99(34):e21659.
doi pubmed pmc - Zonnenberg BA, Neary MP, Duh MS, Ionescu-Ittu R, Fortier J, Vekeman F. Observational study of characteristics and clinical outcomes of Dutch patients with tuberous sclerosis complex and renal angiomyolipoma treated with everolimus. PLoS One. 2018;13(11):e0204646.
doi pubmed pmc - Cai Y, Guo H, Wang W, Li H, Sun H, Shi B, Zhang Y. Assessing the outcomes of everolimus on renal angiomyolipoma associated with tuberous sclerosis complex in China: a two years trial. Orphanet J Rare Dis. 2018;13(1):43.
doi pubmed pmc - Watanabe EH, Coelho FMA, Filho HL, Balbo BEP, Neves P, Franzin FM, Yamauchi FI, et al. The effect of sirolimus on angiomyolipoma is determined by decrease of fat-poor compartments and includes striking reduction of vascular structures. Sci Rep. 2021;11(1):8493.
doi pubmed pmc - Sanfilippo R, Jones RL, Blay JY, Le Cesne A, Provenzano S, Antoniou G, Mir O, et al. Role of chemotherapy, VEGFR inhibitors, and mTOR inhibitors in advanced perivascular epithelioid cell tumors (PEComas). Clin Cancer Res. 2019;25(17):5295-5300.
doi pubmed - Northrup H, Aronow ME, Bebin EM, Bissler J, Darling TN, de Vries PJ, Frost MD, et al. Updated international tuberous sclerosis complex diagnostic criteria and surveillance and management recommendations. Pediatr Neurol. 2021;123:50-66.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.