

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 15, Number 4, August 2024, pages 562-578

Untapped Potential of Poly(ADP-Ribose) Polymerase Inhibitors: Lessons Learned From the Real-World Clinical Homologous Recombination Repair Mutation Testing
Alexandra Lebedevaa, b, Egor Veselovskya, c, Alexandra Kavuna, Ekaterina Belovaa, b, d, Tatiana Grigorevaa, b, e, Pavel Orlovf, Anna Subbotovskayaf, Maksim Shipunovf, Oleg Mashkovg, Fanil Bilalovg, Peter Shatalovh, Andrey Kaprinh, Peter Shegaih, Zhan Diuzhevi, Ochir Migiaevi, Natalya Vytnovai, Vladislav Mileykoa, b, Maxim Ivanova, b, j, k ![]()
aOncoAtlas LLC, Moscow, Russia
bSechenov First Moscow State Medical University, Moscow, Russia
cDepartment of Evolutionary Genetics of Development, Koltzov Institute of Developmental Biology of the Russian Academy of Sciences, Moscow, Russia
dLomonosov Moscow State University, Moscow, Russia
eShemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Moscow, Russia
fThe Federal Research Center for Fundamental and Translational Medicine (NIIECM FRC FTM), Novosibirsk, Russia
gState Budgetary Institution of Healthcare Republican Medical Genetic Center, Ufa, Russia
hNational Medical Research Radiological Centre of the Ministry of Health of the Russian Federation, Obninsk, Russia
iGEMOTEST Laboratory LLC, Moscow, Russia
jMoscow Institute of Physics and Technology, Dolgoprudny, Moscow Region, Russia
kCorresponding Author: Maxim Ivanov, OncoAtlas LLC, Moscow 119049, Russian Federation
Manuscript submitted January 17, 2024, accepted April 29, 2024, published online June 11, 2024
Short title: HRR Testing Uncovers New Actionable Mutations
doi: https://doi.org/10.14740/wjon1820
| Abstract | ▴Top |
Background: Testing for homologous recombination deficiency (HRD) mutations is pivotal to assess individual risk, to proact preventive measures in healthy carriers and to tailor treatments for cancer patients. Increasing prominence of poly(ADP-ribose) polymerase (PARP) inhibitors with remarkable impact on molecular-selected patient survival across diverse nosologies, ingrains testing for BRCA genes and beyond in clinical practice. Nevertheless, testing strategies remain a question of debate. While several pathogenic BRCA1/2 gene variants have been described as founder pathogenic mutations frequently found in patients from Russia, other homologous recombination repair (HRR) genes have not been sufficiently explored. In this study, we present real-world data of routine HRR gene testing in Russia.
Methods: We evaluated clinical and sequencing data from cancer patients who had germline/somatic next-generation sequencing (NGS) HRR gene testing in Russia (BRCA1/2/ATM/CHEK2, or 15 HRR genes). The primary objectives of this study were to evaluate the frequency of BRCA1/2 and non-BRCA gene mutations in real-world unselected patients from Russia, and to determine whether testing beyond BRCA1/2 is feasible.
Results: Data of 2,032 patients were collected from February 2021 to February 2023. Most had breast (n = 715, 35.2%), ovarian (n = 259, 12.7%), pancreatic (n = 85, 4.2%), or prostate cancer (n = 58, 2.9%). We observed 586 variants of uncertain significance (VUS) and 372 deleterious variants (DVs) across 487 patients, with 17.6% HRR-mutation positivity. HRR testing identified 120 (11.8%) BRCA1/2-positive, and 172 (16.9%) HRR-positive patients. With 51 DVs identified in 242 formalin-fixed paraffin-embedded (FFPE), testing for variant origin clarification was required in one case (0.4%). Most BRCA1/2 germline variants were DV (121 DVs, 26 VUS); in non-BRCA1/2 genes, VUS were ubiquitous (53 DVs, 132 VUS). In silico prediction identified additional 4.9% HRR and 1.2% BRCA1/2/ATM/CHEK2 mutation patients.
Conclusions: Our study represents one of the first reports about the incidence of DV and VUS in HRR genes, including genes beyond BRCA1/2, identified in cancer patients from Russia, assessed by NGS. In silico predictions of the observed HRR gene variants suggest that non-BRCA gene testing is likely to result in higher frequency of patients who are candidates for PARP inhibitor therapy. Continuing sequencing efforts should clarify interpretation of frequently observed non-BRCA VUS.
Keywords: Homologous recombination repair; HRR gene mutations; Hereditary breast and ovarian cancer; Pancreatic cancer; Prostate cancer; Founder mutations; PARP inhibitors; Next-generation sequencing
| Introduction | ▴Top |
BRCA1 and BRCA2 play a key role in homologous recombination repair (HRR) of DNA damage and are necessary to maintain genomic integrity in proliferating cells [1]. Germline mutations in at least one of these highly penetrant genes are associated with hereditary breast and ovarian cancer (HBOC) syndrome, and substantially increase the risk of developing various cancer types [2-4], specifically breast, ovarian [5-11], prostate [11], pancreatic [11-14], as well as gastric cancer [15] and potentially melanoma [16]. Moreover, patients can be affected with multiple cancers throughout the lifetime [17]. At the same time, it is becoming clear that not only BRCA1 and BRCA2 are responsible for the hereditary forms of these cancer types. BRCA1/2 interacts with a number of other HRR genes, including ATM, RAD51B/C/D, PALB2, RAD50, NBN, MRE11, CHEK2, BRIP1, BARD1 and the Fanconi anemia proteins [18-20]. Recent evidence suggests that mutations in PALB2 [21-27], ATM [23-26, 28-32], CHEK2 [24-26, 33] and other HRR genes, such as BARD1 [24-26], BRIP1 [27, 32, 34, 35] and RAD51С/RAD51D [24-27, 32, 34, 36, 37] might increase cancer risk. In recent years, poly(ADP-ribose) polymerase (PARP) inhibitors have emerged as a highly effective drug class for patients with BRCA1/2-altered cancers. PARP plays a dominant role in DNA single-strand break repair, and the inhibition of PARP in BRCA1/2-positive tumors leads to the deficiency of single-strand break repair, eventually leading to cell death through a concept known as synthetic lethality [38].
PARP inhibitors such as olaparib, talazoparib, rucaparib, and niraparib have been approved for clinical use in the presence of BRCA1 or BRCA2 variants for the treatment of breast cancer [39, 40], prostate cancer [41, 42], ovarian cancer [43-45], and pancreatic cancer [46]. The presence of variants of HRR genes other than BRCA1 or BRCA2 is an indication for olaparib therapy in patients with prostate cancer [41]. The efficacy of other PARP inhibitors for HRR-mutated prostate cancer has also been studied [47]. For breast [48] and ovarian [49] cancer, olaparib has also been shown to be relatively effective in the presence of variants of several HRR genes.
Next-generation sequencing (NGS)-based approaches give additional benefit via testing of broad target areas of all genes of interest. According to current recommendations, there is a certain group of patients who require molecular genetic testing of BRCA1/2 and other HRR genes. Testing people with a family or personal history of cancer facilitates individualized screening and recommendations to reduce the risk of developing hereditary forms of cancer [50]. According to recent data, approximately 5% of unselected patients with breast cancer [25, 26] and 12-14% of patients with epithelial ovarian cancer [10] might carry germline pathogenic variants in BRCA1/2. A positive family history increases the chance of identifying patients with germline BRCA1/2 variants [51, 52]. However, recent studies show that germline pathogenic variants can also be found in patients who do not have a family history of cancer, and do not meet the criteria for testing [26, 53-55]. According to various studies, about 27-56% of ovarian cancer patients with detected pathogenic BRCA1/2 variants had no reported family history of breast or ovarian cancer [52, 56]. The mean probability of finding a germline BRCA1/2 variant in epithelial ovarian cancer patients without a positive family history for breast and/or ovarian cancer is 6% [52]. This difference may be due to the presence of low-penetrate genes, with an as yet little studied association with cancer risk, or under-researched within the family, where a variant in a high-penetrate gene can be passed through male family members and manifest as cancer at an older age.
The prevalence of pathogenic or uncertain significance (VUS) HRR variants in Russian cancer patients’ population, as well as the age of onset of developing cancer, associated with HBOC syndrome, is still poorly studied. There are previous publications reporting the occurrence of germline variants of the BRCA1/2 genes, as well as ATM in the Russian cancer patient’s population [57-60]. Here, we report the spectrum of BRCA1/2 and HRR gene mutations observed in an unselected real-world cancer patient population in Russia.
| Materials and Methods | ▴Top |
Study population
From February 2021 to February 2023, a total of 2,032 patients with breast, ovarian, prostate, pancreatic, and other types of cancer were rereferred for NGS testing of BRCA1/2 and other HRR genes as a part of routine patient management in five different laboratories (NIIECM FRC FTM, Russia, Novosibirsk - 115 for main analysis and additional 116 for the analysis of geographical distribution of repetitive variants; Republican Medical Genetic Center, Russia, Ufa - 916; GEMOTEST Laboratory LLC, Russia, Moscow - 494; NMRCR, Russia, Moscow - 33; OncoAtlas LLC, Russia, Moscow - 519). Study was approved by Sechenov University IRB, and was conducted in accordance with principles claimed in the Declaration of Helsinki. All participants signed an informed consent, in accordance with local law. All further analyses were based on the archival data that were stored in the database with no current connection to the patients’ identifiers.
Molecular testing
DNA isolation and NGS
DNA was isolated from either whole blood, or formalin-fixed paraffin-embedded (FFPE) tumor samples and subjected to NGS as previously described [61]. In general, QIAamp DNA Blood Kits (Qiagen) was used for DNA isolation from whole blood samples, and GeneRead DNA FFPE Kit (Qiagen) or QIAamp DNA FFPE Tissue Kit (Qiagen) was used for tumor samples. Concentration of extracted DNA as well as concentration of DNA libraries was measured using Qubit Fluorometers with Qubit dsDNA HS and BR Assay Kits (Thermo Fisher Scientific), Bioanalyzer or TapeStation (Agilent Technologies).
DNA sequencing was performed on the Illumina platform (MiSeq or NextSeq550) using Amplicon based NGS kits (IVD certified in Russia): Solo test ABC, Solo test ABC HRR edition. Solo test ABC covers coding regions of ATM, BRCA1, BRCA2 genes, and clinically relevant regions of CHEK2 gene [61]. Solo test ABC HRR edition covers all coding regions of ATM (ENST00000278616), BARD1 (ENST00000260947), BRCA1 (ENST00000471181), BRCA2 (ENST00000544455), BRIP1 (ENST00000259008), CDK12 (ENST00000447079), CHEK1 (ENST00000534070), CHEK2 (ENST00000382580), FANCL (ENST00000402135), PALB2 (ENST00000261584), PPP2R2A (ENST00000315985), RAD51B (ENST00000487270), RAD51C (ENST00000337432), RAD51D (ENST00000590016) and RAD54L (ENST00000371975) genes (transcripts used for variant annotation are denoted in brackets). In order to pass quality control, sequencing data were required to have average depth of 250x and higher (650x for tumor samples), MAPD - 0.5 and lower and sensitivity to detect known BRCA variants of 99.8% and higher [61]. Sequencing datasets failing quality control were not used for retrospective analysis.
Analysis of NGS data and variant interpretation
The analysis of the sequencing data was conducted in accordance with the previously described method [62, 63]. Briefly, reads were mapped on the human genome GRCh37.p13 assembly. Samtools was used for preliminary evaluation of technical characteristics of identified variants, such as variant site coverage depth, observed alternative allele counts, and observed alternative allele frequency. For additional technical annotation of identified variants Mutect2 [64], SiNVICT [65], FreeBayes [66] and SGA [67] were utilized. Variant calls were required to have P-value of 10 × 10-7 and lower after Bonferroni correction. Variants were evaluated based on ACMG [68] and Sherloc guidelines [69]. BRCA Exchange [70] and ClinVar [71] databases were used as main reference sources to classify known variants. Minor allele frequency data were referenced using the 1000 Genomes Project Database [72], the NHLBI GO Exome Sequencing Project [73], and the TOPMED Project [74]. dbSNP database (build 155) was used for variant annotation. Variants detected in tumor samples were classified as germline or somatic according to the previously described algorithm [62]. Variants were classified as somatic in case of 95% and higher probability of somatic origin. In case of 95% and higher probability of germline origin, variant was classified as germline. In other cases, variants were classified as of uncertain origin. Since the analyzed data contained both germline and somatic variants, germline pathogenic and likely pathogenic variants, as well as somatic oncogenic and likely oncogenic variants, were denoted in the manuscript as “deleterious” in order to unify terminology (DV). Variants of uncertain significance, both somatic and germline, are referred to as VUS. CHEK2 variant p.Ile157Thr (rs17879961) was considered as non-deleterious and was not included in the final analysis [75].
| Results | ▴Top |
Patient population
In this study, we retrospectively analyzed sequencing data of 2,032 patients referred for BRCA1/2 and/or broad HRR genes mutation analysis as a part of routine case management from February 2021 to February 2023. Thirty-nine of them had duplicate samples analyzed, mostly genomic DNA (n = 1,335, 64.5%) and tumor DNA (n = 242, 11.7%) samples, resulting in 2,071 samples in total (Table 1). For 492 samples (23.8%), DNA origin was not available for retrospective analysis. Most patients were female (n = 1,177, 57.9%), 83 (4.1%) patients were male, and for other 772 (38%) patients’ sex was not available for retrospective analysis. Median age at testing was 54 years for all genders. Mean age at testing was markedly lower in the females, which can be partly explained by the presence of very young outliers. The majority of patients had breast cancer (n = 715, 35.2%), ovarian cancer (n = 259, 12.7%), pancreatic cancer (n = 85, 4.2%) and prostate cancer (n = 58, 2.9%). Diagnosis for 900 (44.3%) patients was not available for retrospective analysis. Across 1,132 patients with available diagnosis, 15 (1.3%) patients had tumor types that are not routinely tested for HRR gene alterations.
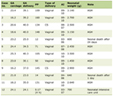 Click to view | Table 1. Clinical and Demographic Characteristics of Patients Included in the Study |
Sequencing results and findings
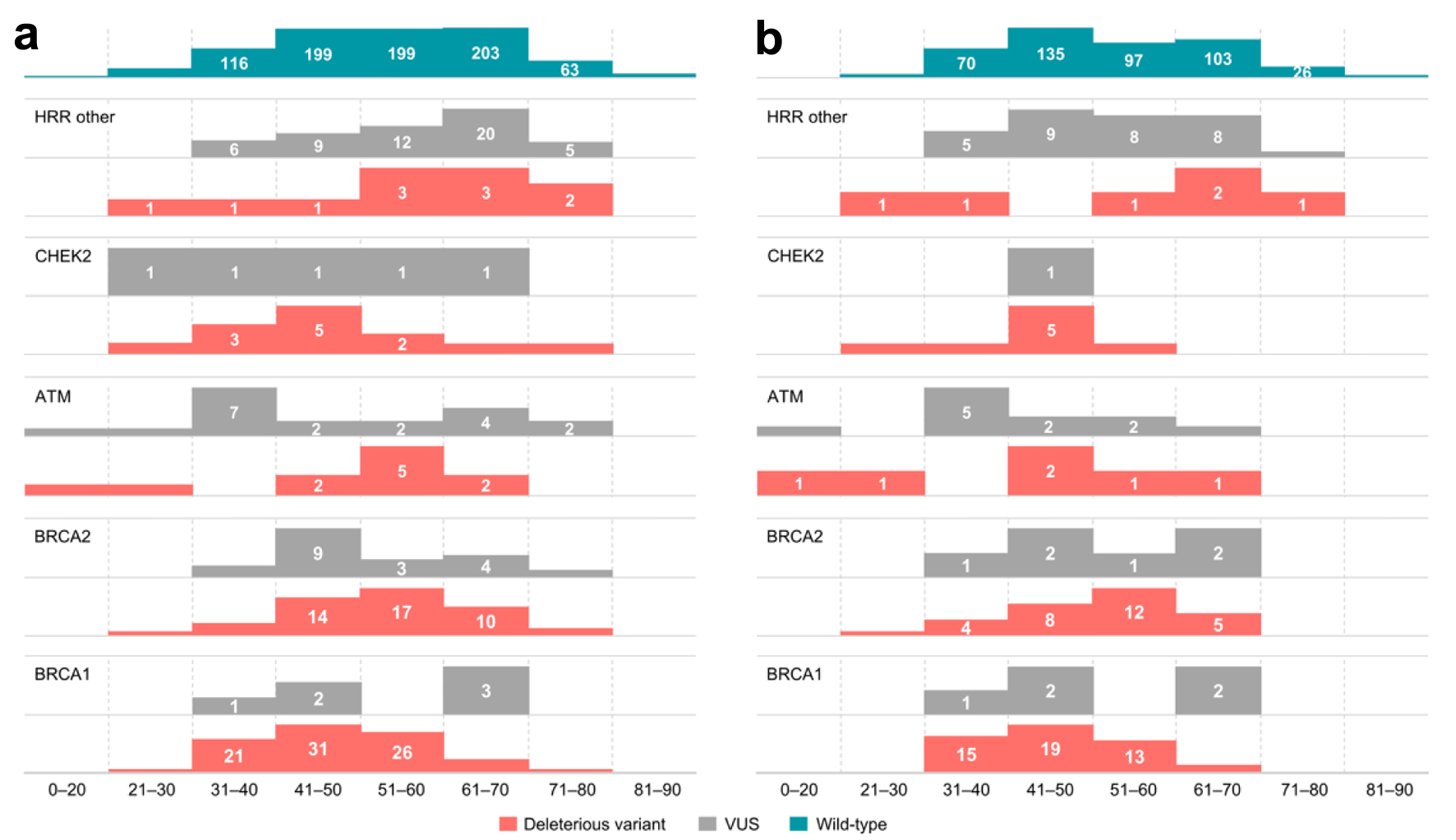
A total of 219 VUS and 372 DVs (including germline pathogenic variants and somatic DVs) were detected with 358 (17.6%) patients carrying at least one DV, 160 (7.8%) patients carrying one or more VUS with no DV found and 1,514 (74.5%) with no DV or VUS detected (Fig. 1). Of all detected variants, 543 (91.8%) were germline (354 DVs, 189 VUS), 16 (2.7%) were somatic (five DVs, 11 VUS), while 32 (5.4%) variants (13 DVs, 19 VUS) could not be reliably classified as germline or somatic (variants of uncertain origin). After filtering out variants that were detected in more than one patient, 318 unique germline, and 31 variants of uncertain origin were observed, while all of the identified somatic variants were unique.
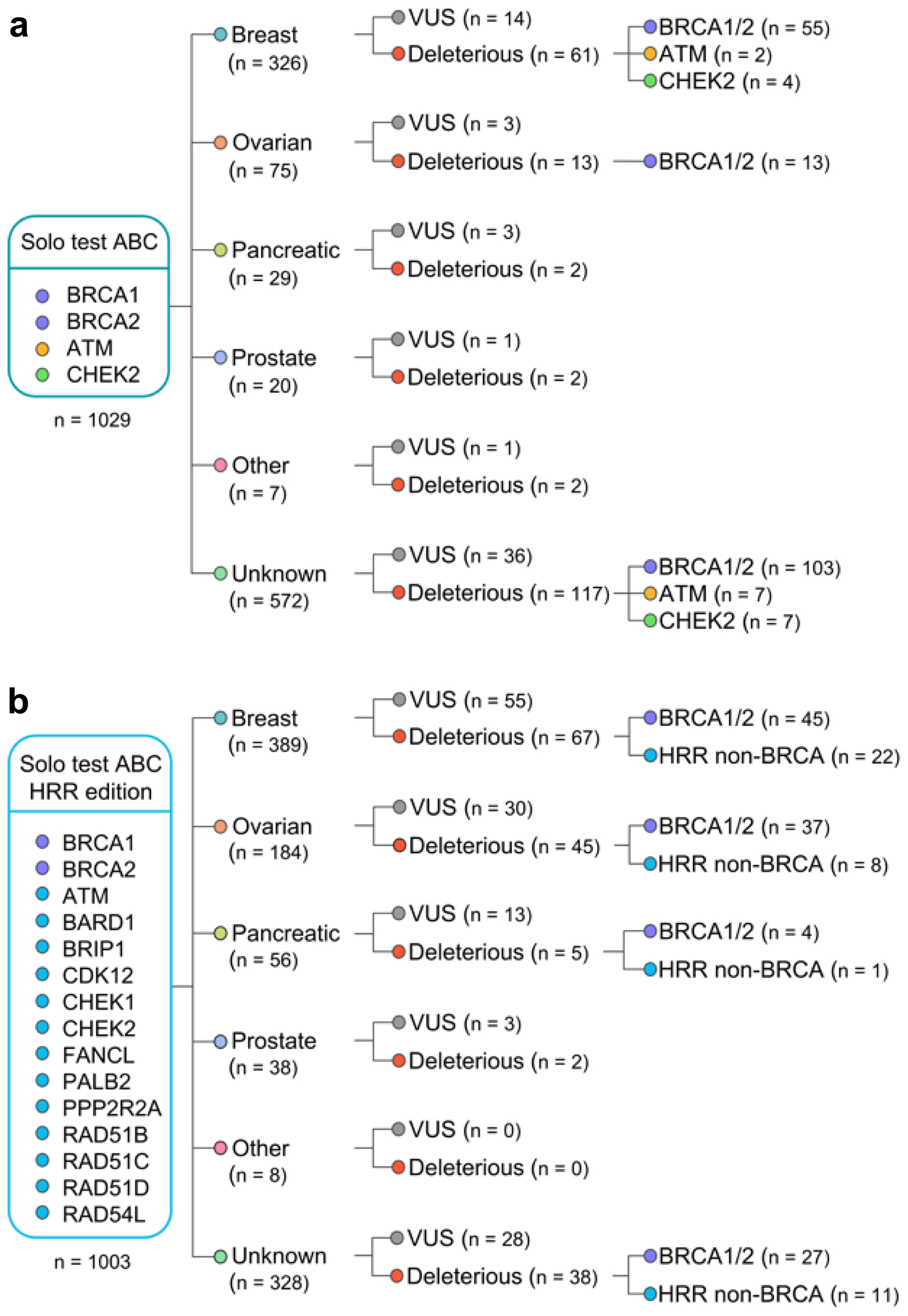 Click for large image | Figure 1. Flow chart with the overview of germline variants identified in the study. Patients were tested with either Solo test ABC (a) or Solo test ABC HRR edition (b). HRR: homologous recombination repair. |
HRR-mutation positive rate defined as the presence of at least one DV in any HRR gene in the whole patient population was 17.6% (95% confidence interval (CI): 15.9-19.2%) (Fig. 2). Across patients analyzed only for BRCA1/2/ATM/CHEK2 genes, HRR-mutation positive rate was 18.5% (95% CI: 16.1-20.9%), whilst for patients analyzed for broad HRR gene panel, it was 16.7% (95% CI: 14.4-19.0%). Across breast cancer patients, 16.9% were HRR-mutation positive (95% CI: 12.7-22.1%), and it was 21.6% for ovarian cancer (95% CI: 7.3-29.8%). Across 715 patients with breast cancer, 98 (13.7%, 95% CI: 11.2-16.2%) were identified with biomarker for PARP inhibitor indication according to FDA drug labeling (germline BRCA1/2 DV). Across a total of 100 BRCA1/2 germline DV identified in breast cancer patients, 11 were identified in tumor samples and 89 in blood samples, and no additional testing was deemed to be necessary to clarify detected variant origin for any breast patient. Across 259 ovarian cancer patients, 53 (20%, 95% CI: 15.5-25.3%) were identified with biomarker for PARP inhibitor indication (germline or somatic BRCA1/2 deleterious mutation) with a total of 53 DVs identified, 50 (94%) of which were germline. In a single ovarian cancer patient, BRCA2 DV identified in a tumor sample was deemed to require verification by blood testing in order to clarify origin of variant. Across 1,003 patients tested for a broad HRR gene panel, 147 were identified with germline DV in one or more genes associated with hereditary cancer syndrome and established recommendations on cancer preventive measures according to NCCN guidelines (NCCN-HC gene panel) with a total of 150 DVs detected (BRCA1/2 - 113, CHEK2 - 13, ATM - 8, RAD51C - 7, BRIP1 - 4, BARD1 - 3, PALB2 - 2), 11 of which were identified in tumor samples. Across 103 tumor samples tested for a broad HRR gene panel, in a single case additional blood testing was deemed to be necessary due to identified DV of uncertain origin in NCCN-HC gene panel.
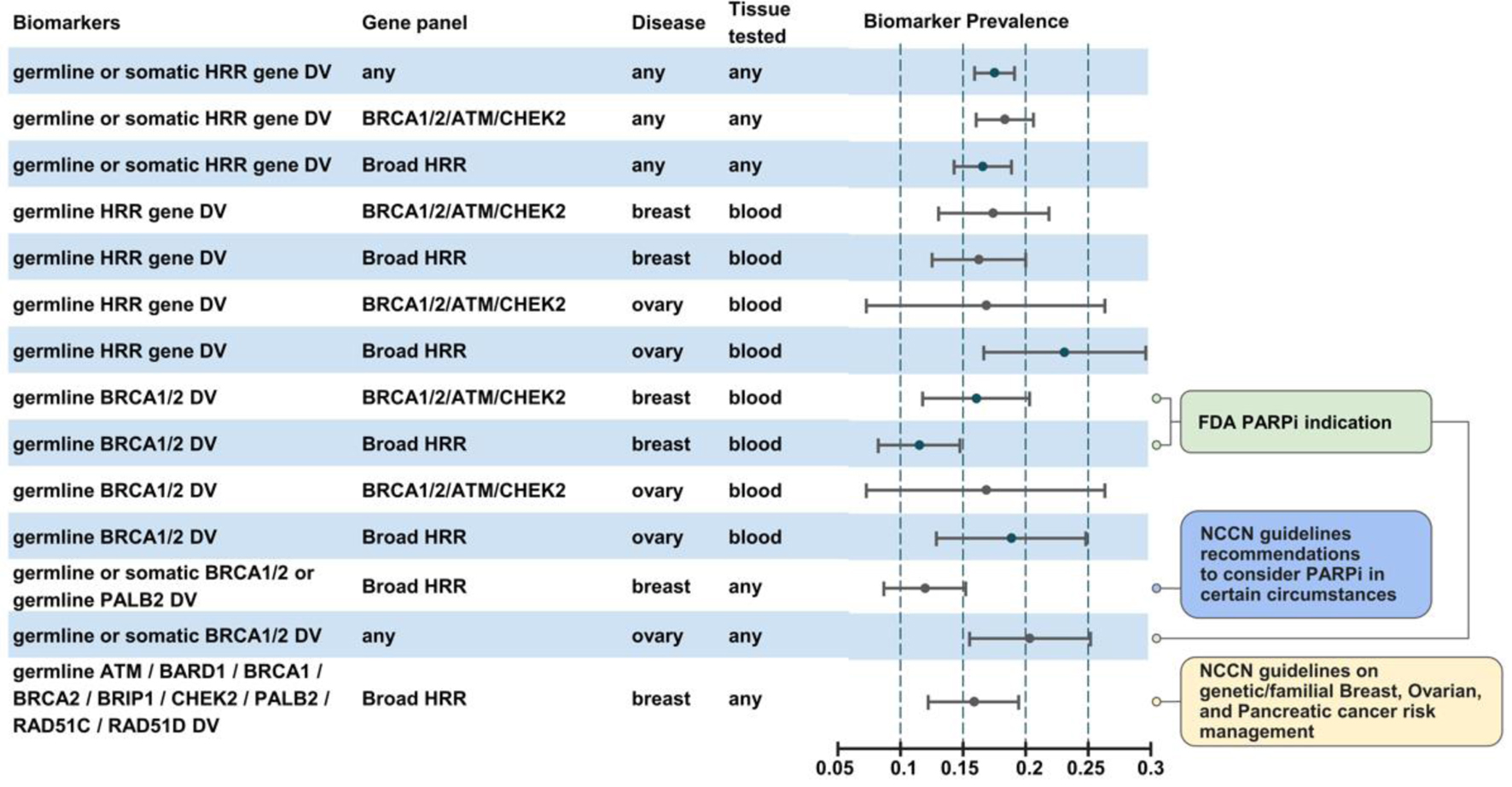 Click for large image | Figure 2. Biomarker prevalence across diverse patient subgroups. |
Among patients with germline DVs, the majority harbored variants in BRCA1/2 genes with a total of 183 (51% of all germline DVs identified) and 109 (29%) variants identified in BRCA1 and BRCA2, respectively. Across patients tested for BRCA1/2, ATM and CHEK2 genes, 24 (7.2%) CHEK2 germline DVs were found and 21 (6.3%) ATM germline DVs were found. Across patients tested for broad HRR gene panel, BRCA1/2 DVs constituted 71% of all DVs identified followed by CHEK2 (8%), ATM (5%) and RAD51C (4%). Germline mutations in other genes were uncommon (17 DVs in total). Of all germline variants detected across all patients including VUS, the majority (n = 357, 66%) were DV, whilst 184 (34%) were VUS. Of 176 unique BRCA1/2 germline variants, 120 were DV, whilst 56 were VUS. On the contrary, among non-BRCA1/2 unique germline variants, the majority (n = 106, 70%) were VUS, whilst 45 (30%) were DV (Fig. 3a). Across patients tested for BRCA1/2, ATM and CHEK2, the majority of identified germline VUS were identified in ATM (n = 42, 40%) followed by BRCA2 (n = 37, 35%), BRCA1 (n = 14, 13%) and CHEK2 (n = 11, 10%). Across patients tested for broad HRR gene panel, the majority of germline VUS were identified in ATM (n = 17, 14%), followed by BRIP1 (n = 23, 19%), CDK12 (n = 16, 13%), BRCA2 (n = 13, 11%), BRCA1 (n = 8, 6%) and CHEK2, RAD54L, PALB2, BARD1 (n = 7, 4% for each).
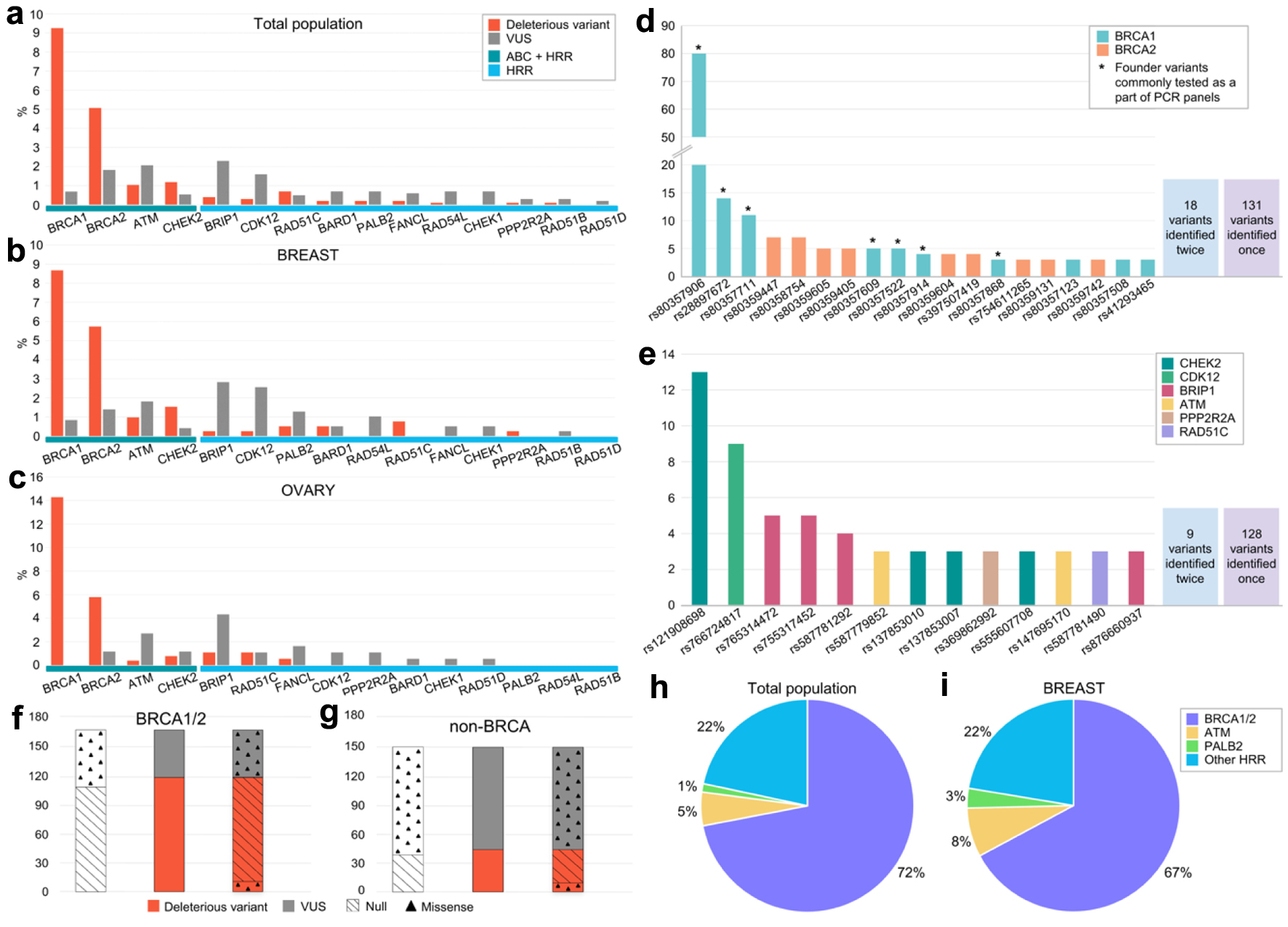 Click for large image | Figure 3. Frequencies of detected germline variants in the analyzed genes. (a-c) Frequencies of DV and VUS variants depending on the gene (a: whole patient population, b: breast cancer patients, c: ovarian cancer patients). (d, e) Frequencies of repetitively identified variants and their distribution by gene (d: BRCA1 and BRCA2, e: non-BRCA1/2). Asterisk indicates variants previously annotated as founder mutations and used to be broadly tested via PCR in Russia. (f, g) Distribution of DV and VUS by type of alteration in BRCA1/2 (f) and non-BRCA (g) genes (null variants comprises nonsense and canonical splice site variants). (h, i) Spectrum of DV in the whole population (h) and across breast cancer patients (i). VUS: variants of uncertain significance; DV: deleterious variants. |
Across 242 tumor samples analyzed, a total of 16 somatic mutations were identified, including 11 VUS and five DVs. None of the patients with somatic DV carried any germline variant, resulting in HRR-mutation positive rate due to somatic mutation of 2.0% (95% CI: 0.6-4.7%) (compared to a total of 19.0% (95% CI: 14.2-24.5%) HRR-mutation positive rate observed across patients with tumor testing). Single somatic DV was identified each in ATM, BRCA1, BRCA2, RAD51D and PPP2R2A. Three somatic VUS were identified in ATM, two each in BRCA1 and BRCA2 and single each in BRIP1, CDK12, RAD54L and BARD1. Across 37 ovarian cancer patients with tumor testing, two DVs (5%, 95% CI: 0.6-18%) and two VUS variants were identified (all in BRCA1/2). Across 67 breast cancer patients with tumor testing, no somatic variants were identified (95% CI: 0.0-5.3%). Across 25 prostate cancer patients with tumor testing, no somatic DVs were identified (95% CI: 0.0-13.7%) and two VUS variants were identified (both in BRCA1/2). Across 10 patients with somatic VUS detected, two carried somatic DV and no one carried germline DV.
Variants of uncertain origin were detected in ATM (n = 8, 25%), BRCA2 (n = 7, 21.8%), BRCA1 (n = 3, 9.4%), CDK12 (n = 3, 9.4%), BARD1 (n = 2, 6.3%), PALB2 (n = 2, 6.3%), and RAD51D (n = 2, 6.3%). Single VUS of uncertain origin were also detected each in CHEK2, BRIP1, FANCL, RAD51B, and RAD54L. Of 31 unique variants of uncertain origin, the majority were classified as VUS (n = 19, 61.3%). Prevalence of DV of uncertain origin across ovarian cancer patients with tumor testing was 2.6% (95% CI: 0.06-13%) (2.6% for VUS of uncertain origin), 0.0% across breast cancer patients (95% CI: 0.0-5.2%) (1.4% for VUS of uncertain origin) and 0.0% across prostate cancer patients (95% CI: 0.0-13.0%) (8% for VUS of uncertain origin). Further testing for clarification of origin of identified in tumor sample DV of uncertain origin due to potential risk for hereditary cancer and, thus, indication for genetic counseling was required for one (0.4%) patient with tumor testing (95% CI: 0.0-2.3%). Further testing for clarification of origin of identified in tumor sample DV of uncertain origin for PARP inhibitor indication was required in 0 (0.0%) patients (95% CI: 0.0-2.4%).
Across a total of 360 unique DVs and VUS detected, 95 (26%) were novel, which were not previously annotated as based on dbSNP database. Each of the novel variants were detected only once. Across the identified novel germline variants, 53 were missense or inframe variants, 33 were nonsense, start-loss or splice site variants while the rest were frameshift variants. Only 37 novel variants (39%) were classified as DV, while the rest were VUS. Seventy-two novel germline variants were detected, 30 (41%) of which were classified as DV. Across all germline DVs, novel variants constituted 8% (22% for VUS). This results in a probability of detecting a novel variant in a single patient of 4.6% (4.5% for breast cancer) and a probability of detecting a novel germline variant of 3.5% (3.3% for breast cancer patients, 4.6% for ovarian, and 5.8% for pancreas). The majority of novel germline DVs were located in BRCA1/2 genes (n = 22, 73%), resulting in a probability of detecting novel germline DV in BRCA1/2 in a single patient of 1% (ATM followed with 0.1%).
Repetitively observed germline variants
A total of 291 germline genetic variants (61 unique alleles), both in BRCA1/2 and non-BRCA1/2 genes, were identified in more than one patient (Fig. 3d, e). A total of 37 unique repetitive variants were observed in BRCA1/2 genes, and 24 were located in non-BRCA1/2 genes. All of these variants were previously annotated in literature and public databases. Among variants in BRCA1/2 genes, the majority were null variants (86.1%). Across non-BRCA1/2 variants, repetitive missense variants were more common (72.7%). The most commonly observed variant was BRCA1 p.Q1777fs (rs80357906), which was found in 80 patients, accounting for 21.8% of all DVs identified across the whole patient population (21.9% and 22.0% across breast cancer and ovarian cancer patients, respectively). Of variants found in three or more samples (n = 19), seven (36.8%) could be potentially identified via standard PCR panels currently used in Russia testing for common BRCA1/2 DV. The most commonly observed variant (n = 13) in non-BRCA1/2 genes was CHEK2 c.444+1G>A (rs121908698).
Given the fact that Russia exhibits significant regional differences in terms of ethnicities, it was decided to compare the frequencies of germline genetic variants obtained from different regions. The largest and comparable groups of patients were derived from the Moscow region (Moscow patients, MP, n = 1,156) and Bashkortostan (Bashkortostan patients, BP, n = 788) (Fig. 4). While Moscow’s population is approximately 90% ethnic Russians, in the Bashkortostan population Russians make up only 48% while the rest of the population is represented by Tatars (27.0%) and Bashkirs (20.4%), according to the 2020 census. As expected, a difference was found between the most common germline variants: only 50% of the eight most frequently occurring variants were found to be the same in both groups. Of eight top variants in the BP, three were not found in the MP. Of these three variants, two were DV in the BRCA2 gene: rs80359447 (n = 7) and rs80358754 (n = 6), representing 9.3% of all DVs found in the BRCA1/2 in the BP. Third unique for the BP variant rs766724817 in the CDK12 gene was classified as VUS, though six out of seven in silico tools predicted deleterious effect of the variant.
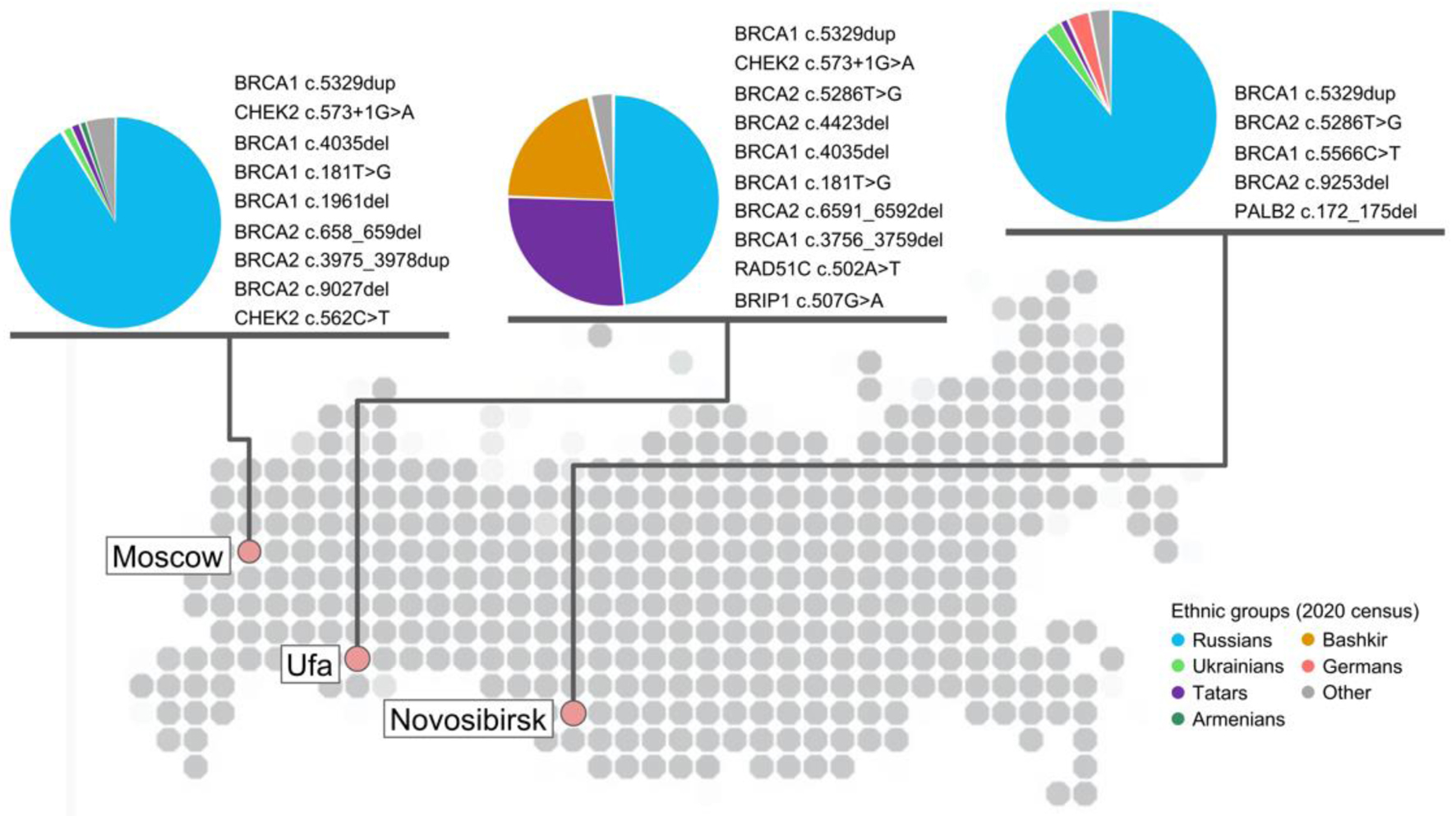 Click for large image | Figure 4. Geographical location of laboratories participated in study with at least 200 samples analyzed and corresponding repetitive germline damaging variants in HRR genes detected in each city in descending order of frequency. Variant was considered as repetitive if it was detected at least two times - for Moscow and Novosibirsk cities; at least three times - for Ufa. Data for Moscow collected based on patients tested with Solo ABC panel; for Ufa and Novosibirsk cities - Solo ABC HRR edition. Ethnic groups with at least 1% representation in each city population are provided for reference and based on the 2020 census in each city. HRR: homologous recombination repair. |
Top eight variants in the MP and in the BP cover 23.1% and 32.5% of all germline variants found in these groups of patients (P-value = 0.0167). Variants unannotated in dbSNP make up 16.6% and 12.2% in the MP and BP groups, respectively (P-value = 0.1751).
Among the MP, 152 (13.1%) had DV in the BRCA1 or BRCA2, and 35 (3.0%) had DV in the non-BRCA genes, while for the BP, the corresponding values were 128 (16.2%) and 34 (4.3%), respectively. In the MP, 75 (6.5%) had no DV, but at least one VUS, and in the BP, 70 (8.9%) had no DV, but at least one VUS. Per each 100 MP and 100 BP patients, 8.9 and 11.5 VUS variants were identified, respectively (P-value = 0.07).
Distribution of findings by tumor type
Germline variants (including DV and VUS) in the analyzed genes were identified across patients with breast (n = 204, 61.3%), ovarian (n = 93, 27.9%), pancreatic (n = 23, 6.9%), prostate cancer (n = 10, 3%), biliary tract (n = 4, 1.2%), as well as uterine cancer (n = 1, 0.3%). Across breast cancer patients, the majority of germline variants were found in BRCA1 (n = 72, 35%) and BRCA2 (n = 52, 35.3%), as well as in ATM (n=20, 9.8%), CHEK2 (n = 14, 6.9%), BRIP1 (n = 12, 5.9%) and CDK12 (n = 11, 5.4%). Germline variants in other genes (including CHEK1, RAD54L, RAD51B, PALB2, RAD51C, BARD1, FANCL, and PPP2R2A) were less common (n < 10). Across patients with ovarian cancer, germline variants in the following genes were the most common: BRCA1 (n = 37, 39.8%), BRCA2 (n = 18, 19.4%), BRIP1 (n = 10, 10.8%), ATM (n = 8, 8.6%), and CHEK2 (n = 5, 5.4%). Across patients affected with pancreatic cancer, BRCA2 (n = 8, 34.8%) variants were the most common, followed by ATM (n = 2), BARD1 (n = 2), and RAD54L (n = 2). Interestingly, no BRCA1 variants were observed in pancreatic cancer patients. Prostate cancer patients harbored germline variants in ATM (n = 2, 25%) and BRIP1 (n = 2, 25%), and variants in other genes (BRCA1, CHEK2, RAD51B, and CDK12) were observed in single cases. All germline variants identified in patients with biliary tract cancer were located in the ATM gene, whilst a uterine cancer patient had a BRCA2 genetic variant. Thus, among breast cancer patients, a total of 81 germline variants (61 unique) in genes other than BRCA1/2 were observed, 38 (31 unique) across ovarian cancer patients, and 14 (all unique) and seven (six unique) across pancreatic and prostate cancer patients, respectively.
Across germline findings, DVs were more common than VUS for patients with breast (135 (66.2%) vs. 69 (33.8%)), ovarian (60 (64.5%) vs. 33 (35.5%)) and biliary tract (4 (100%) vs. 0 (0%)) cancers, whereas patients with pancreatic cancer harbored more VUS than DV (16 (69.6%) vs. 7 (30.4%)). DV and VUS variants were evenly distributed in prostate cancer patients (Fig. 1).
Distribution of germline variants by age
For 1,079 (53.3% of the study population) patients, the information regarding patients’ age was available. Median age of testing for all patients was 54 years. Among tested patients, 823 (76.3%) patients did not have any germline variants, DV or VUS, in any of the analyzed genes. For short, we will refer to these patients as wild type. Of those, 445 patients had breast cancer. Median age of all wild type patients was 54 years, and the median age of wild type breast cancer patients was 52 years. The difference in ages of the whole population and breast cancer patients without any germline variants was statistically insignificant (P = 0.1).
In the whole patient population, the median age of testing across all patients carrying DV and/or VUS variants in any of the HRR genes was 51 years. A difference in the ages of any genetic variant (DV and/or VUS) carriers and wild type patients was statistically significant (P = 0.006). Carriers of any DV were generally younger than wild type patients (median 50 vs. 54 years, P = 0.0004), whereas no statistically significant difference was observed between VUS carriers and wild type patients (median 56 vs. 54 years, P = 0.48). Among all DV carriers, carriers of BRCA1 DV tended to be younger than BRCA2 DV carriers (median, 48 vs. 54 years, P = 0.004). Median age of ATM DV carriers was 51 years, and 57 years for patients with ATM VUS. Median age for CHEK2 DV carriers was 46 years, and 41 years for VUS carriers. Finally, patients with germline DV in other HRR genes had a median age of 57 years, while the median age for carriers of VUS in these genes was 60. Statistically significant difference was found between age distributions of all patients with BRCA1 DV, regardless of tumor type, when compared to HRR wild type patients (P = 0.00002), as BRCA1 DV carriers were younger. Another statistically significant relationship was found between age distributions of BRCA1 DV and HRR DV, as well as VUS carriers (P = 0.02 and P < 0.0001, respectively), as well as when comparing HRR VUS carriers with BRCA2 DV and VUS carriers (P = 0.04 and 0.035, respectively), ATM VUS carriers (P = 0.03), and CHEK2 DV and VUS carriers (P = 0.01 and 0.048, respectively) (Fig. 5a).
 Click for large image | Figure 5. Distribution of germline variants (DV, VUS) by age groups across (a) all patients tested, (b) patients with breast cancer. Data on the age of patients without any identified variants (wild type patients) are presented for reference. VUS: variants of uncertain significance; DV: deleterious variants. |
Separately analysis was performed for breast cancer patients. Median age for breast cancer patients with any germline variants was 48 years. A statistically significant difference was observed between the ages of any DV variant carriers affected with breast cancer and wild type breast cancer patients (median, 49 vs. 52 years, P = 0.018), as well as between VUS carriers and wild type breast cancer patients (median, 44.5 vs. 52 years, P = 0.0001). Median age of breast cancer patients harboring DV were as follows: 44 years for BRCA1, 54 years for BRCA2, 45 years for ATM, 44 years for CHEK2, and 60 years for other HRR genes. Median age for breast cancer carriers of VUS was 48 years for BRCA1, 51 for BRCA2, 37 for ATM, 38 for other HRR genes, including a single CHEK2 VUS carrier, who was 41 years old at the time of testing. In the breast cancer patient population, statistically significant differences were observed between the ages of carriers of BRCA1 DV and ATM VUS when compared to wild type patients (P = 7 × 10-5 and 0.014, respectively). Additionally, a statistically significant difference was observed when comparing BRCA1 and BRCA2 DV carriers (P = 0.004), as well as carriers of VUS in HRR genes other than BRCA1/2, ATM, CHEK2 (P = 0.0067). Finally, a statistically significant interaction was observed between ATM VUS and HRR VUS variant carriers in non-BRCA1/2/ATM/CHEK2 genes (P = 0.033) (Fig. 5b).
A total of 11 patients carried more than a single germline DV. Of those, three patients had double DV in BRCA1/2 genes (two patients had DV in BRCA1 and BRCA2, one patient had two DVs in BRCA2). Three patients harbored concurrent BRCA1/2 and ATM variants (two patients had DV in BRCA2 and ATM, one in BRCA1 and ATM). Two patients harbored DV in BRCA1/2 along with CHEK2. Finally, a single patient had concurrent ATM and CHEK2 DV and a single patient harbored two distinct ATM variants, while another patient had double CHEK2 variants. Furthermore, in addition to DV, 27 patients harbored concurrent VUS variants (one patient had two VUS variants, others had one). Additionally, among those who did not have any DV, 15 patients were found to carry more than a single VUS (two patients had three VUS, others had two). Median age of patients with concurrent germline DV in any of the HRR genes was 52 years. Patients harboring more than a single VUS in any of the analyzed genes had a median age of 61 years, whereas patients with concurrent DV and VUS tended to be younger, with a median age of 45 years. No statistically significant differences were observed between ages of patients with concurrent mutations.
In silico analysis to assess VUS variants
We employed seven in silico tools to predict the effect of identified DV and VUS missense variants, namely, MetaLR [76], VEST [77], CADD [78], FathmmMKL coding [79], MutationTaster [80], SIFT [81] and PROVEAN [82]. Average concordance of prediction results between two tools was 0.69 (range 0.48 - 0.90) with VEST demonstrating highest average concordance with other tools (0.74), whilst MetaLR had the lowest (0.61) (Fig. 6b). CADD, FathmmMKL, MutationTaster and VEST correctly predicted deleterious effect of all BRCA and non-BRCA DVs (Fig. 6c). In line with previous studies [83-86] demonstrating a probability of 95% and higher of pathogenic classification [68] for mutations consistently predicted deleterious by multiple lines of computational tools, all BRCA and non-BRCA missense DVs were predicted to have deleterious effect by six or seven in silico predictors (Fig. 6a). Additionally, 13 BRCA and 39 non-BRCA variants classified as VUS within the general interpretation process (in “Materials and Methods” section) were predicted to have deleterious effects by six or seven in silico predictors. This comprised a total of 52 potentially missed DVs identified in 55 patients (19 - breast cancer patients; eight - ovarian cancer; five - pancreatic cancer; four - prostate cancer) with no other variant classified as DV within the general interpretation process in any gene.
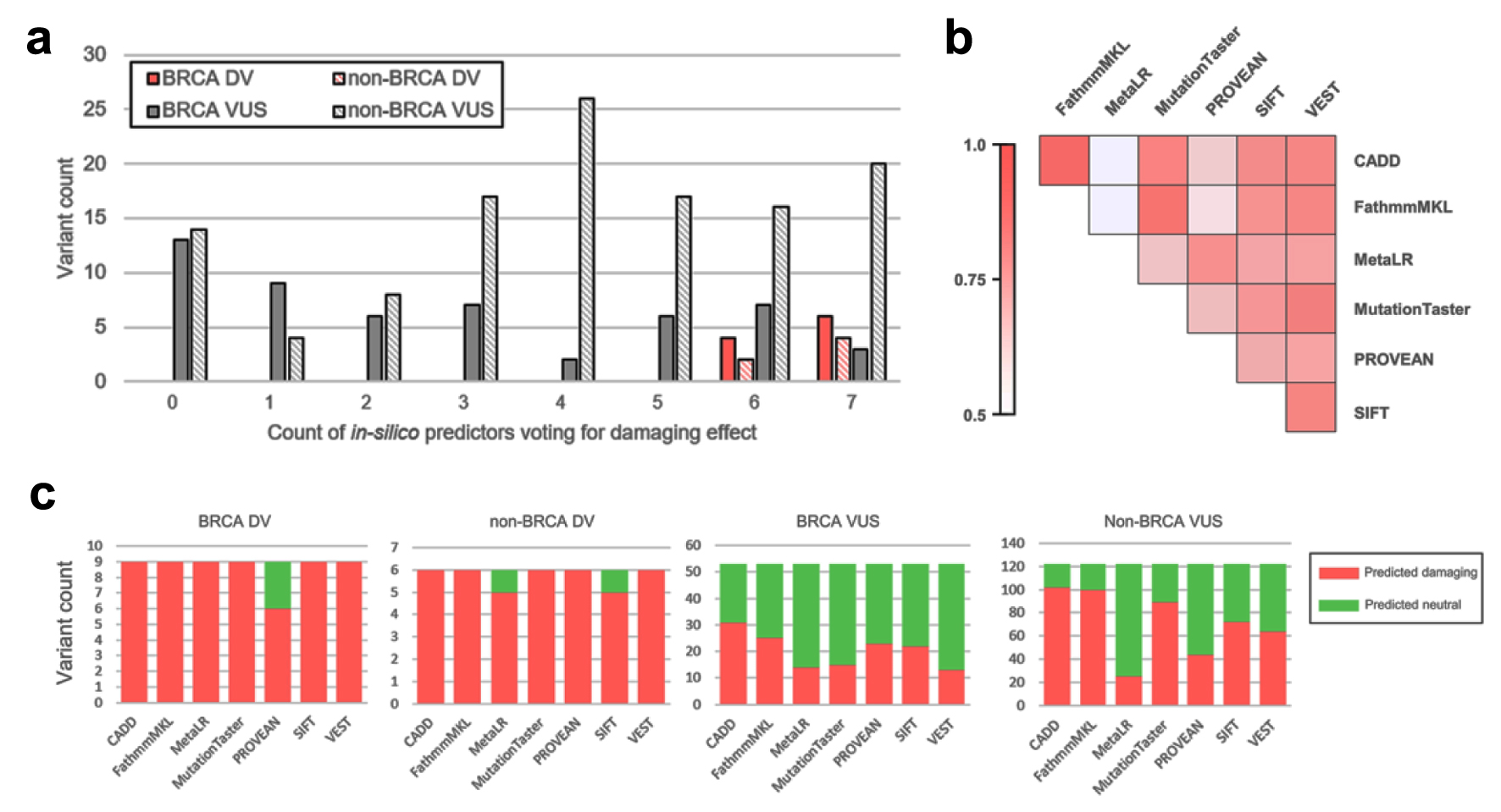 Click for large image | Figure 6. Results of in silico prediction of identified missense variants effect. (a) Distribution of observed VUS and DV by count of in silico tools predicting deleterious effect. (b) Pairwise concordance of in silico prediction algorithms demonstrated on variants identified in study. (c) Per-tool prediction results for BRCA/non-BRCA and VUS/DV. VUS: variants of uncertain significance; DV: deleterious variants. |
| Discussion | ▴Top |
Our study represents one of the first reports about the incidence of DV and VUS variants in HRR genes identified in breast, ovarian, pancreatic and prostate cancer patients from Russia, assessed by NGS. Across 2,032 patients tested, 24% harbored HRR gene variants, of which BRCA1/2 variants (16.4%) were the most common, followed by ATM (3%) and CHEK2 (1.7%) variants. The incidence of HRR gene variants is reported to be high among patients with HOBC type of cancer; however, it can vary significantly among different countries or some ethnic groups due to the founder effect [87]. Our results are in line with those previously published. Frequency of BRCA1/2 pathogenic variants is similar with previously reported for breast cancer patients from Eastern Sicily [88] or Poland and Ukraine [89] (9% vs. 15.1% vs. 14% in our study), and correlates well with reported for ovarian cancer patients from Brazil [90] or Poland and Ukraine [89] (20.8% vs. 23.8% vs. 20.4% in our study). However, in non-BRCA1/2-mutated breast cancer Hispanic Americans [91], the frequency of other HRR pathogenic variants is practically the same (4.5% vs. 3.9% in our study) and rare incidence of PALB2 pathogenic variants coincides with reported for breast and ovarian cancer patients from Poland (1.5% vs. 0.2%) [92]. A high frequency of BRCA1 VUS or pathogenic variants has previously been reported in the Russian population of patients with breast cancer, and the results are consistent with those obtained in our study (7.4% vs. 9%) [93]. Observed frequency of pathogenic CHEK2 germline variants (1.5%) is close to the known for the Baltic (4%) and Finnish (3.7%) populations of breast cancer patients [94, 95], as well as in worldwide data (1.4%) [25].
In 2020, American Society of Clinical Oncology (ASCO) recommended that all women diagnosed with epithelial ovarian cancer should have germline testing for BRCA1/2 and other ovarian cancer susceptibility genes followed by tumor testing for patients who do not carry a germline pathogenic or likely pathogenic variant [96]. Further studies in contradiction demonstrated higher cost-efficiency of tumor testing triage for germline testing strategy for patients with epithelial ovarian cancer [97]. For patients with prostate cancer, parallel germline and somatic testing is recommended for patients who may benefit from PARP inhibitors [98]. The same ambiguity of testing strategies comes to the fore for breast cancer patients as the latest NCCN guidelines recommend somatic BRCA testing as useful in certain circumstances, though, PARP inhibitors are currently not FDA approved as treatment for these mutations [99]. Moreover, some breast cancer patients would require tumor testing for ESR1, HER2 and PIK3CA mutations proposing usage of larger panels including BRCA genes to exclude the need of additional testing in future. Overall, this demonstrates high uncertainty of optimal germline and tumor testing sequencing for patients who both may benefit from targeted therapy and may require to rule out heredity of disease. Our study included real-world data of both germline and somatic testing employing both BRCA1/2/ATM/CHEK2 genes panel and broad HRR genes panel. HRR genes testing has almost the same efficiency within germline and somatic testing frameworks and resulted in 16% and 17% of HRR-mutation positive cases, respectively (P-value = 0.19). The same percentage of positive cases was seen in a total study population including cases when tested material was not reported. BRCA1/2/ATM/CHEK2 genes testing resulted in contrasting efficiency with 24%, 13% and 18% of positive cases for somatic testing, germline testing and for total study population, respectively. What’s more important, across 51 DV mutations identified in 242 FFPE samples tested, further testing for variant origin clarification was required only in a single case (0.4%), whilst other 50 variants could be identified as somatic or germline with high confidence based solely on FFPE sequencing data (in “Materials and Methods” section). Overall, this demonstrates high efficiency of the tumor testing triage for germline testing strategy.
In our study, BRCA1/2 variants were the most common across observed germline DVs (80.5% across all patients and 71.5% across patients tested for broad HRR panel). Patients with prostate and pancreatic cancer were characterized by higher prevalence of somatic mutation, higher prevalence of BRCA2 germline DVs compared to BRCA1 germline DV and higher prevalence of non-BRCA germline DVs. This difference in mutation frequencies can be attributed to the phenotype variability of hereditary cancer syndromes associated with defects in HR [100, 101], as well as heterogeneity of the analyzed patient population and differences in panels used. When analyzing patients with available age, breast cancer patients harboring germline variants in any of the genes were younger, and this difference was statistically significant. The peak incidence of breast cancer in BRCA1/2 DV carriers was between 41 and 60 years at the time of testing, consistent with currently available evidence [5, 102].
The majority of BRCA1/2 germline variants identified in our patient cohort were DV, while for the non-BRCA1/2 genes, the opposite dynamic was observed, with more VUS being identified (Fig. 3a-c). Several recurrent variants were observed, both in BRCA1/2 and non-BRCA1/2 genes (Fig. 3d, e). Of those, 17 (48.6%) BRCA1/2 and four (18.2%) non-BRCA1/2 variants have been previously described as founder mutations, commonly observed in Eastern European, European, Russian, or other ethnically similar populations [87, 103-114]. When comparing our results with standard BRCA1/2 PCR assays, only 42.6% of patients who were found to harbor DV in either BRCA1, or BRCA2 in our study, could be identified as BRCA1/2-positive via PCR panels routinely employed in clinical practice in Russia. Early studies concluded that up to 90% of all BRCA1/2 mutations found in the Russian Slavic population are represented by common founder variants, thus, PCR may be an efficient method for BRCA mutation analysis [115]. Nevertheless, recent studies indicated the need of full-length BRCA1/2 sequencing [59] which is consistent with our data. Additionally, several VUS were identified more than once, predominantly in non-BRCA1/2 genes (68.2% VUS among non-BRCA1/2 genes vs. 5.6% VUS in BRCA1/2). This difference can be attributed to the colossal efforts toward BRCA1/2 variant classification that have resulted in clarification of clinical significance of a number of variants, including missense variants, and accumulation of data in specific knowledge bases [116-119]. At the same time, the clinical significance of variants occurring in other HRR genes is less widely studied, possibly due to the rarity of these events and the fact that testing for non-BRCA1/2 variants has been a relatively recent addition to the clinical practice [120, 121].
Studies show that therapeutic benefit of PARP inhibitors is not limited to DV in BRCA1/2 genes, as alterations in other genes and transcriptomic signatures have been linked to this drug class [41, 122-126]. Therefore, identification of DV in HRR genes is of utmost importance for therapeutic management of breast, ovarian, pancreatic and prostate cancers. The clarification of clinical significance of non-BRCA1/2 HRR gene variants is crucial for selecting patients who would potentially benefit from PARP inhibitor therapy. Since several non-BRCA1/2 variants were identified more than once in our study, it might be possible that a fraction of patients who in fact are candidates for PARP inhibitor therapy, fail to receive it.
Acute shortage of literature data for variant interpretation comes to the fore as broad HRR gene panels are introduced into clinical practice. First of all, in contrast to variants observed in BRCA1/2, missense variants are more common in non-BRCA HRR genes rather than null variants (including variants resulting in premature translation termination and canonical splice site variants). Considering that variants with population frequency of 0.3% and higher are defined as neutral, a total of 550 variants required interpretation in our study. This included 343 variants located in BRCA1/2 and 207 located in HRR genes other than BRCA1/2. Among BRCA1/2 variants, 272 (79%) were null variants and, thus, did not require exhaustive literature search for variant interpretation, while only 128 (47%) variants in non-BRCA HRR genes were null variants. Consequently, this resulted in higher incidence of VUS variants identified in non-BRCA HRR genes (13.7 and 2.4 VUS variants identified per each 100 patients in non-BRCA and BRCA genes, respectively). Employing in silico prediction tools, we identified 13 BRCA1/2 and 39 non-BRCA VUS variants with high (95% and more) probability of deleterious effect due to concordant prediction by multiple lines of computation tools, which, however, cannot be interpreted as DV due to lack of functional or case-control studies described in literature. This potentially may result in 50 patients tested employing a broad HRR gene panel missing their positive result due to lack of data available for variant interpretation, accounting for additional 4.9% potentially positive patients in addition to 16.5% patients with DV. In contrast, across patients tested employing BRCA1/2/ATM/CHEK2 panel, these calculations result only in 1.2% potentially positive patients in addition to 18.8% patients with DV. Further sequencing efforts may reduce this gap in BRCA vs. non-BRCA variant annotation in literature.
Taken together, our results highlight the need for continuous research aiming at evaluation of clinical significance of non-BRCA1/2 VUS. Furthermore, our results highlight the need for using NGS as a method of choice for BRCA1/2 germline variant detection, especially in PCR-negative patients. Expanding testing beyond BRCA1/2 variants might be reasonable, especially for BRCA1/2 wild type patients. Finally, we demonstrate that our laboratory-developed panels can be efficiently used for the mutational analysis of BRCA1/2 and other HRR genes in patients with various cancer types.
The primary limitation of our study is that the complete data on patients’ sex, age and tumor type were only available for a fraction of patients, and information on family history of cancer was available for none of the patients. Additionally, partly due to the fact that a large number of laboratories were involved in the testing, no follow-up information was available for any of the patients, making it impossible to make any conclusions on how the testing had influenced that patients’ lives. Furthermore, since only a fraction of patients were tested for variants in non-BRCA1/2/ATM/CHEK2 genes, the incidence of variants in other HRR genes was tested in a smaller patient population.
In conclusion, our study demonstrates frequencies of BRCA1/2 and other HRR gene variants and explores the relationship between variant carriers and various patient characteristics in a large real-world unselected cancer patient cohort. We describe potential founder variants that were observed not only in BRCA1/2, but also in other HRR genes when tested with our own laboratory-developed panels. Finally, our study highlights the significant advantages of NGS in comparison with PCR for HRR variant detection.
Acknowledgments
None to declare.
Financial Disclosure
The work was partially performed using the equipment of the Center for Collective Use “Proteomic Analysis”, supported by funding from the Ministry of Science and Higher Education of the Russian Federation (No. 075-15-2021-691).
Conflict of Interest
Alexandra Lebedeva, Egor Veselovsky, Alexandra Kavun, Ekaterina Belova, Tatiana Grigoreva, Vladislav Mileyko, Maxim Ivanov are employees of OncoAtlas LLC. Zhan Diuzhev, Ochir Migiaev, Natalya Vytnova are employees of GEMOTEST Laboratory LLC. Other co-authors have no conflict of interest to disclose.
Informed Consent
Patients provided informed consent before participation in this retrospective study. All further analyses were based on the archival data stored in the database with no current connection to the patients’ identifiers.
Author Contributions
Tatiana Grigoreva, Pavel Orlov, Anna Subbotovskaya, Maksim Shipunov, Oleg Mashkov, Fanil Bilalov, Peter Shatalov, Andrey Kaprin, Peter Shegai, Zhan Diuzhev, Ochir Migiaev, and Natalya Vytnova carried out the studies, participated in collecting data. Alexandra Lebedeva, Egor Veselovsky, Alexandra Kavun, Ekaterina Belova, and Maxim Ivanov performed data analysis and drafted the manuscript. Vladislav Mileyko and Maxim Ivanov provided administrative support. All authors contributed to manuscript revision, read, and approved the submitted version.
Data Availability
Data supporting the findings of this study are available from the corresponding author on reasonable request.
| References | ▴Top |
- Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2011;12(1):68-78.
doi pubmed pmc - Easton DF, Bishop DT, Ford D, Crockford GP. Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. Am J Hum Genet. 1993;52(4):678-701.
pubmed pmc - Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66-71.
doi pubmed - Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559):789-792.
doi pubmed - Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, Jervis S, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317(23):2402-2416.
doi pubmed - Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117-1130.
doi pubmed pmc - Mavaddat N, Peock S, Frost D, Ellis S, Platte R, Fineberg E, Evans DG, et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst. 2013;105(11):812-822.
doi pubmed - Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet. 1998;62(3):676-689.
doi pubmed pmc - van der Kolk DM, de Bock GH, Leegte BK, Schaapveld M, Mourits MJ, de Vries J, van der Hout AH, et al. Penetrance of breast cancer, ovarian cancer and contralateral breast cancer in BRCA1 and BRCA2 families: high cancer incidence at older age. Breast Cancer Res Treat. 2010;124(3):643-651.
doi pubmed - Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25(11):1329-1333.
doi pubmed pmc - Breast Cancer Linkage Consortium. Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91(15):1310-1316.
doi pubmed - Ferrone CR, Levine DA, Tang LH, Allen PJ, Jarnagin W, Brennan MF, Offit K, et al. BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma. J Clin Oncol. 2009;27(3):433-438.
doi pubmed pmc - Lerner N, Blei F, Bierman F, Johnson L, Piomelli S. Chelation therapy and cardiac status in older patients with thalassemia major. Am J Pediatr Hematol Oncol. 1990;12(1):56-60.
doi pubmed - Kim DH, Crawford B, Ziegler J, Beattie MS. Prevalence and characteristics of pancreatic cancer in families with BRCA1 and BRCA2 mutations. Fam Cancer. 2009;8(2):153-158.
doi pubmed pmc - Buckley KH, Niccum BA, Maxwell KN, Katona BW. Gastric Cancer Risk and Pathogenesis in BRCA1 and BRCA2 Carriers. Cancers (Basel). 2022;14(23):5953.
doi pubmed pmc - Gumaste PV, Penn LA, Cymerman RM, Kirchhoff T, Polsky D, McLellan B. Skin cancer risk in BRCA1/2 mutation carriers. Br J Dermatol. 2015;172(6):1498-1506.
doi pubmed pmc - Oshi M, Yamada A, Kimura A, Kataoka T, Kobayashi N, Ichikawa Y, Yamanaka S, et al. A case of BRCA2-pathogenic variant breast cancer with metachronous endometrial cancer and pancreatic cancer. World J Oncol. 2023;14(4):309-315.
doi pubmed pmc - Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016;27(8):1449-1455.
doi pubmed - Lupo B, Trusolino L. Inhibition of poly(ADP-ribosyl)ation in cancer: old and new paradigms revisited. Biochim Biophys Acta. 2014;1846(1):201-215.
doi pubmed - Yamamoto H, Hirasawa A. Homologous recombination deficiencies and hereditary tumors. Int J Mol Sci. 2021;23(1):348.
doi pubmed pmc - Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkas K, Roberts J, Lee A, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371(6):497-506.
doi pubmed pmc - Casadei S, Norquist BM, Walsh T, Stray S, Mandell JB, Lee MK, Stamatoyannopoulos JA, et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. 2011;71(6):2222-2229.
doi pubmed pmc - Grant RC, Al-Sukhni W, Borgida AE, Holter S, Kanji ZS, McPherson T, Whelan E, et al. Exome sequencing identifies nonsegregating nonsense ATM and PALB2 variants in familial pancreatic cancer. Hum Genomics. 2013;7(1):11.
doi pubmed pmc - Breast Cancer Association C, Mavaddat N, Dorling L, Carvalho S, Allen J, Gonzalez-Neira A, Keeman R, et al. Pathology of tumors associated with pathogenic germline variants in 9 breast cancer susceptibility genes. JAMA Oncol. 2022;8(3):e216744.
doi pubmed pmc - Breast Cancer Association Consortium, Dorling L, Carvalho S, Allen J, Gonzalez-Neira A, Luccarini C, Wahlstrom C, et al. Breast cancer risk genes - association analysis in more than 113,000 women. N Engl J Med. 2021;384(5):428-439.
doi pubmed pmc - Hu C, Hart SN, Gnanaolivu R, Huang H, Lee KY, Na J, Gao C, et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N Engl J Med. 2021;384(5):440-451.
doi pubmed pmc - Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, Bernards SS, et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 2016;2(4):482-490.
doi pubmed pmc - Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, Byrd P, et al. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97(11):813-822.
doi pubmed - Cavaciuti E, Lauge A, Janin N, Ossian K, Hall J, Stoppa-Lyonnet D, Andrieu N. Cancer risk according to type and location of ATM mutation in ataxia-telangiectasia families. Genes Chromosomes Cancer. 2005;42(1):1-9.
doi pubmed - Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, Gallinger S, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2(1):41-46.
doi pubmed pmc - Angele S, Falconer A, Edwards SM, Dork T, Bremer M, Moullan N, Chapot B, et al. ATM polymorphisms as risk factors for prostate cancer development. Br J Cancer. 2004;91(4):783-787.
doi pubmed pmc - Kurian AW, Hughes E, Handorf EA, Gutin A, Allen B, Hartman AR, Hall MJ. Breast and ovarian cancer penetrance estimates derived from germline multiple-gene sequencing results in women. JCO Precis Oncol. 2017;1:1-12.
doi pubmed - Stolarova L, Kleiblova P, Janatova M, Soukupova J, Zemankova P, Macurek L, Kleibl Z. CHEK2 germline variants in cancer predisposition: stalemate rather than checkmate. Cells. 2020;9(12):2675.
doi pubmed pmc - Lilyquist J, LaDuca H, Polley E, Davis BT, Shimelis H, Hu C, Hart SN, et al. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol Oncol. 2017;147(2):375-380.
doi pubmed pmc - Weber-Lassalle N, Hauke J, Ramser J, Richters L, Gross E, Blumcke B, Gehrig A, et al. BRIP1 loss-of-function mutations confer high risk for familial ovarian cancer, but not familial breast cancer. Breast Cancer Res. 2018;20(1):7.
doi pubmed pmc - Song H, Dicks E, Ramus SJ, Tyrer JP, Intermaggio MP, Hayward J, Edlund CK, et al. Contribution of germline mutations in the RAD51B, RAD51C, and RAD51D genes to ovarian cancer in the population. J Clin Oncol. 2015;33(26):2901-2907.
doi pubmed pmc - Yang X, Song H, Leslie G, Engel C, Hahnen E, Auber B, Horvath J, et al. Ovarian and breast cancer risks associated with pathogenic variants in RAD51C and RAD51D. J Natl Cancer Inst. 2020;112(12):1242-1250.
doi pubmed pmc - Lau CH, Seow KM, Chen KH. The molecular mechanisms of actions, effects, and clinical implications of PARP inhibitors in epithelial ovarian cancers: a systematic review. Int J Mol Sci. 2022;23(15):8125.
doi pubmed pmc - Robson ME, Tung N, Conte P, Im SA, Senkus E, Xu B, Masuda N, et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician's choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann Oncol. 2019;30(4):558-566.
doi pubmed pmc - Litton JK, Hurvitz SA, Mina LA, Rugo HS, Lee KH, Goncalves A, Diab S, et al. Talazoparib versus chemotherapy in patients with germline BRCA1/2-mutated HER2-negative advanced breast cancer: final overall survival results from the EMBRACA trial. Ann Oncol. 2020;31(11):1526-1535.
doi pubmed pmc - de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, Chi KN, et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med. 2020;382(22):2091-2102.
doi pubmed - Abida W, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, Sautois B, et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol. 2020;38(32):3763-3772.
doi pubmed pmc - Banerjee S, Moore KN, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2021;22(12):1721-1731.
doi pubmed - Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, Colombo N, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949-1961.
doi pubmed pmc - Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154-2164.
doi pubmed - Kindler HL, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, et al. Overall survival results from the POLO trial: a phase iii study of active maintenance Olaparib versus placebo for germline BRCA-mutated metastatic pancreatic cancer. J Clin Oncol. 2022;40(34):3929-3939.
doi pubmed pmc - Abida W, Campbell D, Patnaik A, Shapiro JD, Sautois B, Vogelzang NJ, Voog EG, et al. Non-BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration-resistant prostate cancer: analysis from the phase II TRITON2 study. Clin Cancer Res. 2020;26(11):2487-2496.
doi pubmed pmc - Tung NM, Robson ME, Ventz S, Santa-Maria CA, Nanda R, Marcom PK, Shah PD, et al. TBCRC 048: phase II study of olaparib for metastatic breast cancer and mutations in homologous recombination-related genes. J Clin Oncol. 2020;38(36):4274-4282.
doi pubmed - Hodgson DR, Dougherty BA, Lai Z, Fielding A, Grinsted L, Spencer S, O'Connor MJ, et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br J Cancer. 2018;119(11):1401-1409.
doi pubmed pmc - Pujol P, Barberis M, Beer P, Friedman E, Piulats JM, Capoluongo ED, Garcia Foncillas J, et al. Clinical practice guidelines for BRCA1 and BRCA2 genetic testing. Eur J Cancer. 2021;146:30-47.
doi pubmed - Kurian AW, Abrahamse P, Ward KC, Hamilton AS, Deapen D, Berek JS, Hoang L, et al. Association of family cancer history with pathogenic variants in specific breast cancer susceptibility genes. JCO Precis Oncol. 2021;5:1853-1859.
doi pubmed pmc - Arts-de Jong M, de Bock GH, van Asperen CJ, Mourits MJ, de Hullu JA, Kets CM. Germline BRCA1/2 mutation testing is indicated in every patient with epithelial ovarian cancer: A systematic review. Eur J Cancer. 2016;61:137-145.
doi pubmed - Singer CF, Tan YY, Muhr D, Rappaport C, Gschwantler-Kaulich D, Grimm C, Polterauer S, et al. Association between family history, mutation locations, and prevalence of BRCA1 or 2 mutations in ovarian cancer patients. Cancer Med. 2019;8(4):1875-1881.
doi pubmed pmc - Momozawa Y, Iwasaki Y, Parsons MT, Kamatani Y, Takahashi A, Tamura C, Katagiri T, et al. Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls. Nat Commun. 2018;9(1):4083.
doi pubmed pmc - Tung N, Lin NU, Kidd J, Allen BA, Singh N, Wenstrup RJ, Hartman AR, et al. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J Clin Oncol. 2016;34(13):1460-1468.
doi pubmed pmc - Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J, Dobrovic A, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol. 2012;30(21):2654-2663.
doi pubmed pmc - Bogdanova N, Cybulski C, Bermisheva M, Datsyuk I, Yamini P, Hillemanns P, Antonenkova NN, et al. A nonsense mutation (E1978X) in the ATM gene is associated with breast cancer. Breast Cancer Res Treat. 2009;118(1):207-211.
doi pubmed - Kechin A, Boyarskikh U, Barinov A, Tanas A, Kazakova S, Zhevlova A, Khrapov E, et al. A spectrum of BRCA1 and BRCA2 germline deleterious variants in ovarian cancer in Russia. Breast Cancer Res Treat. 2023;197(2):387-395.
doi pubmed - Sokolenko AP, Sokolova TN, Ni VI, Preobrazhenskaya EV, Iyevleva AG, Aleksakhina SN, Romanko AA, et al. Frequency and spectrum of founder and non-founder BRCA1 and BRCA2 mutations in a large series of Russian breast cancer and ovarian cancer patients. Breast Cancer Res Treat. 2020;184(1):229-235.
doi pubmed - Solodskikh SA, Panevina AV, Gryaznova MV, Gureev AP, Serzhantova OV, Mikhailov AA, Maslov AY, et al. Targeted sequencing to discover germline variants in the BRCA1 and BRCA2 genes in a Russian population and their association with breast cancer risk. Mutat Res. 2019;813:51-57.
doi pubmed - Ivanov M, Ivanov M, Kasianov A, Rozhavskaya E, Musienko S, Baranova A, Mileyko V. Novel bioinformatics quality control metric for next-generation sequencing experiments in the clinical context. Nucleic Acids Res. 2019;47(21):e135.
doi pubmed pmc - Lebedeva A, Shaykhutdinova Y, Seriak D, Ignatova E, Rozhavskaya E, Vardhan D, Manicka S, et al. Incidental germline findings during molecular profiling of tumor tissues for precision oncology: molecular survey and methodological obstacles. J Transl Med. 2022;20(1):29.
doi pubmed pmc - Ivanov M, Laktionov K, Breder V, Chernenko P, Novikova E, Telysheva E, Musienko S, et al. Towards standardization of next-generation sequencing of FFPE samples for clinical oncology: intrinsic obstacles and possible solutions. J Transl Med. 2017;15(1):22.
doi pubmed pmc - McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297-1303.
doi pubmed pmc - Kockan C, Hach F, Sarrafi I, Bell RH, McConeghy B, Beja K, Haegert A, et al. SiNVICT: ultra-sensitive detection of single nucleotide variants and indels in circulating tumour DNA. Bioinformatics. 2017;33(1):26-34.
doi pubmed - Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing (Version 2). arXiv:2012.
doi - Simpson JT, Durbin R. Efficient de novo assembly of large genomes using compressed data structures. Genome Res. 2012;22(3):549-556.
doi pubmed pmc - Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-424.
doi pubmed pmc - Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho YY, Kobayashi Y, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med. 2017;19(10):1105-1117.
doi pubmed pmc - Cline MS, Liao RG, Parsons MT, Paten B, Alquaddoomi F, Antoniou A, Baxter S, et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018;14(12):e1007752.
doi pubmed pmc - Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062-D1067.
doi pubmed pmc - Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061-1073.
doi pubmed pmc - Fu W, O'Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493(7431):216-220.
doi pubmed pmc - Kowalski MH, Qian H, Hou Z, Rosen JD, Tapia AL, Shan Y, Jain D, et al. Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet. 2019;15(12):e1008500.
doi pubmed pmc - Ivanov M, Sharova M, Olsen A, Lebedeva A, Ignatova E, Mouse G, Mileyko V. Letter to the Editor: CHEK2 I157T - Pluto Among Numerous Low-Risk Genetic Factors Requiring Discharge From a Range of Pathogenic Variants? J Natl Compr Canc Netw. 2022;20(2):xxv.
doi pubmed - Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32(8):894-899.
doi pubmed pmc - Carter H, Douville C, Stenson PD, Cooper DN, Karchin R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics. 2013;14 Suppl 3:S3.
doi pubmed pmc - Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886-D894.
doi pubmed pmc - Shihab HA, Rogers MF, Gough J, Mort M, Cooper DN, Day IN, Gaunt TR, et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015;31(10):1536-1543.
doi pubmed pmc - Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361-362.
doi pubmed - Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812-3814.
doi pubmed pmc - Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745-2747.
doi pubmed pmc - Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet. 2006;7:61-80.
doi pubmed - Khafizov K, Ivanov MV, Glazova OV, Kovalenko SP. Computational approaches to study the effects of small genomic variations. J Mol Model. 2015;21(10):251.
doi pubmed - Leong IU, Stuckey A, Lai D, Skinner JR, Love DR. Assessment of the predictive accuracy of five in silico prediction tools, alone or in combination, and two metaservers to classify long QT syndrome gene mutations. BMC Med Genet. 2015;16:34.
doi pubmed pmc - Ernst C, Hahnen E, Engel C, Nothnagel M, Weber J, Schmutzler RK, Hauke J. Performance of in silico prediction tools for the classification of rare BRCA1/2 missense variants in clinical diagnostics. BMC Med Genomics. 2018;11(1):35.
doi pubmed pmc - Ferla R, Calo V, Cascio S, Rinaldi G, Badalamenti G, Carreca I, Surmacz E, et al. Founder mutations in BRCA1 and BRCA2 genes. Ann Oncol. 2007;18(Suppl 6):vi93-98.
doi pubmed - Stella S, Vitale SR, Martorana F, Massimino M, Pavone G, Lanzafame K, Bianca S, et al. Mutational Analysis of BRCA1 and BRCA2 Genes in Breast Cancer Patients from Eastern Sicily. Cancer Manag Res. 2022;14:1341-1352.
doi pubmed pmc - Nguyen-Dumont T, Karpinski P, Sasiadek MM, Akopyan H, Steen JA, Theys D, Hammet F, et al. Genetic testing in Poland and Ukraine: should comprehensive germline testing of BRCA1 and BRCA2 be recommended for women with breast and ovarian cancer? Genet Res (Camb). 2020;102:e6.
doi pubmed pmc - Cotrim DP, Ribeiro ARG, Paixao D, de Queiroz Soares DC, Jbili R, Pandolfi NC, Cezana C, et al. Prevalence of BRCA1 and BRCA2 pathogenic and likely pathogenic variants in non-selected ovarian carcinoma patients in Brazil. BMC Cancer. 2019;19(1):4.
doi pubmed pmc - Weitzel JN, Neuhausen SL, Adamson A, Tao S, Ricker C, Maoz A, Rosenblatt M, et al. Pathogenic and likely pathogenic variants in PALB2, CHEK2, and other known breast cancer susceptibility genes among 1054 BRCA-negative Hispanics with breast cancer. Cancer. 2019;125(16):2829-2836.
doi pubmed pmc - Kluska A, Balabas A, Piatkowska M, Czarny K, Paczkowska K, Nowakowska D, Mikula M, et al. PALB2 mutations in BRCA1/2-mutation negative breast and ovarian cancer patients from Poland. BMC Med Genomics. 2017;10(1):14.
doi pubmed pmc - Iyevleva AG, Suspitsin EN, Kroeze K, Gorodnova TV, Sokolenko AP, Buslov KG, Voskresenskiy DA, et al. Non-founder BRCA1 mutations in Russian breast cancer patients. Cancer Lett. 2010;298(2):258-263.
doi pubmed - Pavlovica K, Irmejs A, Noukas M, Palover M, Kals M, Tonisson N, Metspalu A, et al. Spectrum and frequency of CHEK2 variants in breast cancer affected and general population in the Baltic states region, initial results and literature review. Eur J Med Genet. 2022;65(5):104477.
doi pubmed - Kuusisto KM, Bebel A, Vihinen M, Schleutker J, Sallinen SL. Screening for BRCA1, BRCA2, CHEK2, PALB2, BRIP1, RAD50, and CDH1 mutations in high-risk Finnish BRCA1/2-founder mutation-negative breast and/or ovarian cancer individuals. Breast Cancer Res. 2011;13(1):R20.
doi pubmed pmc - Konstantinopoulos PA, Norquist B, Lacchetti C, Armstrong D, Grisham RN, Goodfellow PJ, Kohn EC, et al. Germline and somatic tumor testing in epithelial ovarian cancer: ASCO guideline. J Clin Oncol. 2020;38(11):1222-1245.
doi pubmed pmc - Kwon JS, Tinker AV, Santos J, Compton K, Sun S, Schrader KA, Karsan A. Germline testing and somatic tumor testing for BRCA1/2 pathogenic variants in ovarian cancer: What is the optimal sequence of testing? JCO Precis Oncol. 2022;6:e2200033.
doi pubmed pmc - Cheng HH, Sokolova AO, Schaeffer EM, Small EJ, Higano CS. Germline and somatic mutations in prostate cancer for the clinician. J Natl Compr Canc Netw. 2019;17(5):515-521.
doi pubmed - Gradishar WJ, Moran MS, Abraham J, Abramson V, Aft R, Agnese D, Allison KH, et al. NCCN Guidelines(R) insights: breast cancer, Version 4.2023. J Natl Compr Canc Netw. 2023;21(6):594-608.
doi pubmed - Castera L, Harter V, Muller E, Krieger S, Goardon N, Ricou A, Rousselin A, et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet Med. 2018;20(12):1677-1686.
doi pubmed - Astiazaran-Symonds E, Kim J, Haley JS, Kim SY, Rao HS, Genetics Center R, Carey DJ, et al. A Genome-First Approach to Estimate Prevalence of Germline Pathogenic Variants and Risk of Pancreatic Cancer in Select Cancer Susceptibility Genes. Cancers (Basel). 2022;14(13):3257.
doi pubmed pmc - Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst. 2002;94(18):1365-1372.
doi pubmed - Gorodetska I, Serga S, Lahuta T, Ostapchenko L, Demydov S, Khranovska N, Skachkova O, et al. Prevalence of two BRCA1 mutations, 5382insC and 300T > G, in ovarian cancer patients from Ukraine. Fam Cancer. 2017;16(4):471-476.
doi pubmed - Lhotova K, Stolarova L, Zemankova P, Vocka M, Janatova M, Borecka M, Cerna M, et al. Multigene panel germline testing of 1333 Czech patients with ovarian cancer. Cancers (Basel). 2020;12(4):956.
doi pubmed pmc - Kluska A, Balabas A, Paziewska A, Kulecka M, Nowakowska D, Mikula M, Ostrowski J. New recurrent BRCA1/2 mutations in Polish patients with familial breast/ovarian cancer detected by next generation sequencing. BMC Med Genomics. 2015;8:19.
doi pubmed pmc - Kowalik A, Siolek M, Kopczynski J, Krawiec K, Kalisz J, Zieba S, Kozak-Klonowska B, et al. BRCA1 founder mutations and beyond in the Polish population: A single-institution BRCA1/2 next-generation sequencing study. PLoS One. 2018;13(7):e0201086.
doi pubmed pmc - Janavicius R. Founder BRCA1/2 mutations in the Europe: implications for hereditary breast-ovarian cancer prevention and control. EPMA J. 2010;1(3):397-412.
doi pubmed pmc - Kluz T, Jasiewicz A, Marczyk E, Jach R, Jakubowska A, Lubinski J, Narod SA, et al. Frequency of BRCA1 and BRCA2 causative founder variants in ovarian cancer patients in South-East Poland. Hered Cancer Clin Pract. 2018;16:6.
doi pubmed pmc - Lawniczak M, Jakubowska A, Bialek A, Lubinski J, Jaworska-Bieniek K, Kaczmarek K, Starzynska T. BRCA1 founder mutations do not contribute to increased risk of gastric cancer in the Polish population. Hered Cancer Clin Pract. 2016;14:3.
doi pubmed pmc - Chessa L, Piane M, Magliozzi M, Torrente I, Savio C, Lulli P, De Luca A, et al. Founder effects for ATM gene mutations in Italian Ataxia Telangiectasia families. Ann Hum Genet. 2009;73(Pt 5):532-539.
doi pubmed - Mitui M, Bernatowska E, Pietrucha B, Piotrowska-Jastrzebska J, Eng L, Nahas S, Teraoka S, et al. ATM gene founder haplotypes and associated mutations in Polish families with ataxia-telangiectasia. Ann Hum Genet. 2005;69(Pt 6):657-664.
doi pubmed - [Frequency of CHEK2 gene mutations in patients with breast cancer from the Republic of Bashkortostan]. Mol Biol (Mosk). 2014;48(1):55-61.
pubmed - Sokolenko AP, Rozanov ME, Mitiushkina NV, Sherina NY, Iyevleva AG, Chekmariova EV, Buslov KG, et al. Founder mutations in early-onset, familial and bilateral breast cancer patients from Russia. Fam Cancer. 2007;6(3):281-286.
doi pubmed - Bogdanova N, Enssen-Dubrowinskaja N, Feshchenko S, Lazjuk GI, Rogov YI, Dammann O, Bremer M, et al. Association of two mutations in the CHEK2 gene with breast cancer. Int J Cancer. 2005;116(2):263-266.
doi pubmed - Imyanitov Y, Belyaev A, Shcherbakov A, Bershteyn L, Bessonov A, Krivorotko P, Gorodnova T, et al. Presence of BRCA1 AND BRCA2 in healthy women and men: dna testing, diagnostic activities and cancer prevention. In Problems in oncology. Autonomous non-profit scientific and medical organization - Questions of Oncology. 2017;63(2):190-198.
doi - Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro AN, Tavtigian SV, Couch FJ, Breast Cancer Information Core Steering C. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet. 2004;75(4):535-544.
doi pubmed pmc - Parsons MT, Tudini E, Li H, Hahnen E, Wappenschmidt B, Feliubadalo L, Aalfs CM, et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: An ENIGMA resource to support clinical variant classification. Hum Mutat. 2019;40(9):1557-1578.
doi pubmed pmc - Thomassen M, Blanco A, Montagna M, Hansen TV, Pedersen IS, Gutierrez-Enriquez S, Menendez M, et al. Characterization of BRCA1 and BRCA2 splicing variants: a collaborative report by ENIGMA consortium members. Breast Cancer Res Treat. 2012;132(3):1009-1023.
doi pubmed - Toland AE, Brody LC, BIC Steering Committee. Lessons learned from two decades of BRCA1 and BRCA2 genetic testing: the evolution of data sharing and variant classification. Genet Med. 2019;21(7):1476-1480.
doi pubmed pmc - Brandao RD, Mensaert K, Lopez-Perolio I, Tserpelis D, Xenakis M, Lattimore V, Walker LC, et al. Targeted RNA-seq successfully identifies normal and pathogenic splicing events in breast/ovarian cancer susceptibility and Lynch syndrome genes. Int J Cancer. 2019;145(2):401-414.
doi pubmed pmc - Nielsen SM, Eccles DM, Romero IL, Al-Mulla F, Balmana J, Biancolella M, Bslok R, et al. Genetic testing and clinical management practices for variants in non-BRCA1/2 Breast (and Breast/Ovarian) cancer susceptibility genes: an international survey by the evidence-based network for the interpretation of germline mutant alleles (ENIGMA) clinical working group. JCO Precis Oncol. 2018;2:1-42.
doi pubmed pmc - Gruber JJ, Afghahi A, Timms K, DeWees A, Gross W, Aushev VN, Wu HT, et al. A phase II study of talazoparib monotherapy in patients with wild-type BRCA1 and BRCA2 with a mutation in other homologous recombination genes. Nat Cancer. 2022;3(10):1181-1191.
doi pubmed pmc - Reiss KA, Mick R, O'Hara MH, Teitelbaum U, Karasic TB, Schneider C, Cowden S, et al. Phase II study of maintenance rucaparib in patients with platinum-sensitive advanced pancreatic cancer and a pathogenic germline or somatic variant in BRCA1, BRCA2, or PALB2. J Clin Oncol. 2021;39(22):2497-2505.
doi pubmed - Pujade-Lauraine E, Brown J, Barnicle A, Wessen J, Lao-Sirieix P, Criscione SW, du Bois A, et al. Homologous recombination repair gene mutations to predict olaparib plus bevacizumab efficacy in the first-line ovarian cancer PAOLA-1/ENGOT-ov25 trial. JCO Precis Oncol. 2023;7:e2200258.
doi pubmed pmc - Tung NM, Robson ME, Ventz S, Santa-Maria CA, Marcom PK, Nanda R, Shah PD, et al. TBCRC 048: A phase II study of olaparib monotherapy in metastatic breast cancer patients with germline or somatic mutations in DNA damage response (DDR) pathway genes (Olaparib Expanded). Journal of Clinical Oncology. 2020;38(15_suppl):1002-1002.
doi - Oshi M, Gandhi S, Wu R, Asaoka M, Yan L, Yamada A, Yamamoto S, et al. Development of a novel BRCAness score that predicts response to PARP inhibitors. Biomark Res. 2022;10(1):80.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.