

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 15, Number 5, October 2024, pages 809-824

SNX4 Is Correlated With Immune Infiltration and Prognosis in Clear Cell Renal Cell Carcinoma
Yu Meng Chaia, c, Zhong Bao Zhoua, c, Run Ze Liua, Yuan Shan Cuib, d, Yong Zhanga, d
aDepartment of Urology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100070, China
bDepartment of Urology, The Affiliated Yantai Yuhuangding Hospital of Qingdao University, Yantai, Shandong, China
cThese authors contributed equally to this article.
dCorresponding Author: Yong Zhang, Department of Urology, Beijing Tiantan Hospital, Capital Medical University, Beijing 100070, China; Yuan Shan Cui, Department of Urology, The Affiliated Yantai Yuhuangding Hospital of Qingdao University, Yantai, Shandong, China
Manuscript submitted April 6, 2024, accepted June 18, 2024, published online July 18, 2024
Short title: SNX4 Expression and Prognosis in ccRCC
doi: https://doi.org/10.14740/wjon1868
| Abstract | ▴Top |
Background: Clear cell renal cell carcinoma (ccRCC) is known as the most common and malignant histologic subtype of renal carcinoma. Sorting nexin 4 (SNX4) plays a regulatory role in recycling from endosomes to the plasma membrane and promotes autophagosome assembly and transport, which may exert the cancerous growth and progression. This study aimed to assess the biological role of SNX4 in ccRCC and their clinical association via public biological data platforms combined with experimental verification.
Methods: In our study, we analyzed the mRNA and protein expression of SNX4 in ccRCC under different clinicopathological characteristics through The Cancer Genome Atlas (TCGA), Human Protein Atlas (HPA) and Clinical Proteomic Tumor Analysis Consortium (CPTAC) databases. We used the Gene Expression Profiling Interactive Analysis (GEPIA) platform to conduct the survival analysis and figure out the immune cell infiltration level under different expression levels of SNX4 combined with Tumor Immune Estimation Resource (TIMER) database. Furthermore, we predicted competing endogenous RNA (ceRNA) regulatory network using TargetScan, miRDB, starBase and miRTarBase online databases. We totally collected six paired ccRCC tissues and adjacent tissues and applied quantitative real-time polymerase chain reaction (qRT-PCR) and western blot (WB) to detect the expression of SNX4 in the collected clinical specimens.
Results: The mRNA and protein expression level of SNX4 was significantly lower in ccRCC than those in normal tissues. The results proposed that lower SNX4 was expressed in patients with higher histologic grade and in male patients. Kaplan-Meier analysis demonstrated that lower mRNA expression level of SNX4 was correlated with poorer prognosis. SNX4 had positive correlation with immune cell infiltrating levels and programmed cell death-ligand 1 (PD-L1) expression. Furthermore, we constructed the SNX4/miR-221-3p/miR-222-3p/DHRS4-AS1 axis, which may be the underlying ceRNA interaction network. Finally, we verified the reduced expression of SNX4 in ccRCC by qRT-PCR and WB.
Conclusion: The expression of SNX4 in ccRCC was lower than adjacent tissues and its downregulated expression was associated with poor prognosis of ccRCC patients. SNX4 may exert critical roles in the tumorigenesis, development and migration of ccRCC via various mechanisms.
Keywords: SNX4; ccRCC; Bioinformatics; Prognosis
| Introduction | ▴Top |
Kidney cancer represents approximately 2% of all oncological diagnoses and deaths worldwide [1]. Clear cell renal cell carcinoma (ccRCC) is the most common and malignant histologic subtype of renal carcinoma with a growing global incidence [2]. Men are more easily affected than women (a 2:1 ratio of new diagnoses) [1]. Localized RCC can mostly be treated with partial or radical nephrectomy. However, patients with RCC are often asymptomatic and a subset of patients are diagnosed with unresectable or metastatic forms. Despite recent advances in treatment options, the long-term prognosis still remains poor, which requires systemic therapies and is associated with high mortality. Targeted therapy has been widely developed, but treatment response is uncertain and treatment resistance in long-term use is inevitable and most patients eventually progress. However, an unprecedented number of drugs are approved to use, and advances in treatment landscape have lifted the survival curves for this disease markedly in recent years.
Large-scale tumor transcriptome analyses demonstrated that ccRCC is highly immune-infiltrated among all solid tumor types [3, 4], which is proven to be correlated with poor outcomes following nephrectomy. Growing evidence suggests that immune checkpoint blockade (ICB) therapy and combination regimens have emerged as a promising treatment option for advanced ccRCC patients in the era of biological targeted therapy [5-8]. Studies have presented that tumor mutation burden (TMB) and programmed cell death-ligand 1 (PD-L1) level can barely predict the therapeutic response in ccRCC [9, 10]. Increasing researches have given a new insight into the genomic and molecular characterizations of metastatic ccRCC and altered pathways and cellular processes [11, 12]. Multiple gene mutations and their interactions contribute to the pathogenesis and may also play a role in immune therapeutic response. Thus, it is necessary to explore the underlying molecular mechanisms of ccRCC initiation and progression, which may provide beneficial effect for treatment.
Sorting nexins (SNXs) are a family of membrane-associated proteins transporting proteins throughout the cells and balancing protein homeostasis. Functioning as sorting proteins, SNXs proteins play a pivotal role in endosomal signaling and multiple endosomal transport pathways [13]. Biological disorders in SNXs are involved in the imbalance of intracellular metabolisms, impaired homeostatic responses, and underlying disease occurrence, such as cancerous and neoplastic diseases. Increasing studies reported that SNX family is involved in tumor occurrence, development, metastasis and drug resistance [14]. Sorting nexin 4 (SNX4) contains both PI(3)P binding PX domain and BAR domain, and exerts regulatory roles in recycling from endosomes to the plasma membrane reported in yeast and mammalian cells. Anton et al’s experiments illustrated that SNX4 complexes act as positive regulators of autophagy that promote autophagosome assembly kinetics by coordinating ATG9A trafficking within the endocytic network [15]. Research further showed that SNX4-assisted vacuolar targeting pathway was responsible for the autophagic response by controlling the degradation of both positive and negative functional transcriptional regulators of ATG [16]. SNX-BARs exist as homo- and hetero-dimers, and SNX4 is involved to construct functional heterodimers with either SNX7 or SNX30, which is related to tubulovesicular endocytic membranes [17].
However, it is still unclear whether endosomal sorting by SNX4 proteins gives rise to tumorigenesis and development of ccRCC. In this study, we investigated its expression level and its correlation with clinical data by using different databases. We also assessed the immune infiltration level in ccRCC using the online databases and further explored the underlying mediated noncoding RNA (ncRNA), covering microRNAs (miRNAs) and long noncoding RNAs (lncRNAs), and built a competing endogenous RNA (ceRNA) network [18].
| Materials and Methods | ▴Top |
The Cancer Genome Atlas (TCGA) and the Gene Expression Omnibus (GEO) databases
TCGA database [19], which is freely accessible, provides large-scale cancer genomics data sets covering over 30 tumor types. We investigated RNA sequencing (RNA-seq) of ccRCC and corresponding clinical pathology data from TCGA database.
The GEO database [20], supported by National Center for Biotechnology Information (NCBI), is a publicly available database that contains a large number of gene expression profiles regarding various diseases. Since the data for gene expression data are public available, it is unnecessary to require approval from the Ethics Committee.
The Human Protein Atlas (HPA), UALCAN, and Clinical Proteomic Tumor Analysis Consortium (CPTAC)
The HPA [21, 22] is initiated to provide protein expression information in cells, tissues, and organs using an integration of various omics technologies. All the data resources are open access to allow exploration of the distribution of the proteins across all major tissues and organs in the human body. UALCAN [23-25] is visual web portal that analyzes the correlations between RNA-sequence and patient clinical data on TCGA dataset. Proteomics data were accessed and downloaded from the CPTAC portal [26] to acquire ccRCC and normal tissue samples used in this study. Samples from CPTAC were analyzed by mass spectrometry (MS)-based proteomics methods. Constituent proteins and expression values were identified and quantified. By the above approach, we obtained SNX4 protein expression data from CPTAC in UALCAN portal tools.
LinkedOmics database
LinkedOmics database [27, 28] contains multi-omics data and clinical data for 32 cancer types and integrates MS-based global proteomics data generated by the CPTAC on selected TCGA tumor samples. Three analysis modules were developed in the LinkedOmics web application. LinkFinder module was adopted to acquire statistical test results in the form of volcano plots and data for 50 top-ranking attributes are visualized in heatmaps. Spearman correlation test was used for analysis.
Tumor Immune Estimation Resource (TIMER) database
TIMER [29, 30] is a web-accessible resource to allow further explorations of the clinical impact of different immune infiltrates in the different tumor microenvironments. In our study, this website was used to characterize the interrelationship and association between abundance of immune infiltrates and SNX4 gene expression in ccRCC.
The Gene Expression Profiling Interactive Analysis (GEPIA)
The GEPIA [31] serves as a web server for gene expression profiling based on TCGA samples as well as survival analysis and correlative analyses by different tumor characteristics. We applied GEPIA to evaluate the correlation of SNX4 expression level and immune cell infiltration level and explore the association between SNX4 expression and immune cells markers.
Univariable, multivariable Cox regression analysis and logistic regression analysis
We generated a Kaplan-Meier survival curve to assess the impact of SNX4 expression on the prognosis of ccRCC patients. Additionally, we conducted univariable and multivariable Cox regression analyses to investigate whether SNX4, along with various clinical features (such as age, gender, T stage, N stage, M stage, pathological stage, and histologic grade), could serve as independent factors influencing the prognosis of individuals with ccRCC. Logistic regression analysis was applied to all variables to compute the odds ratio (OR) and 95% confidence interval (CI), aiming to identify and elucidate the risk factors associated with ccRCC.
Establishment of the nomogram
We employed the “rms” package in R (version 3.6.3) to develop a nomogram incorporating independently identified risk factors for predicting the overall survival (OS) of ccRCC patients at 1, 3, and 5 years. Furthermore, calibration curves were constructed to assess the nomogram’s accuracy.
Prediction and analysis of upstream miRNAs and corresponding lncRNAs of SNX4
The prediction of upstream miRNAs that could potentially bind to SNX4 was conducted using online databases TargetScan [32], miRDB [33, 34], starBase [35] and miRTarBase [36]. Venn diagram was drawn by bioinformatics to confirm the mutual predictive target genes. To visualize the results, a miRNA-SNX4 regulatory network was constructed using cytoscape software. The upstream lncRNAs of microRNAs were predicted using starBase database and the expression correlation between lncRNAs and hsa-miR-221-3p/hsa-miR-222-3p or SNX4 in ccRCC was also detected using starBase database.
Patient and clinical samples
Eleven ccRCC and their adjacent tissue samples were randomly selected from the patients who underwent radical nephrectomy. Ethical approval was granted from the Ethics Committee of Beijing Tiantan Hospital, Capital Medical University (approval number: KY2024-005-01). The study was conducted in compliance with the ethical standards of the responsible institution on human subjects as well as with the Helsinki Declaration.
RNA extraction, reverse transcription and quantitative real-time polymerase chain reaction (qRT-PCR)
We extracted total RNA from freshly frozen ccRCC tissues and their adjacent normal tissues using SteadyPure Quick RNA Extraction Kit (Accurate Biology, China) and then conducted reverse transcription using Evo M-MLV RT Mix Kit (Accurate Biology, China). Next, we carried out qRT-PCR by using SYBR® Green Premix Pro Taq HS qPCR Kit and Rox Reference Dye (Accurate Biology, China). The housekeeping gene GAPDH was used as the internal reference, and the relative expression level of the target gene was calculated by the 2-DDCT calculation method. Three multiple holes were set for each experiment.
Western blot (WB)
Total protein was extracted from the collected tissues using a standardized protein extraction buffer containing protease and phosphatase inhibitors. The concentration of extracted proteins was determined using BCA assay. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was prepared to separate proteins, which were transferred onto a nitrocellulose membrane through a semi-dry transfer method. The membrane was then blocked to prevent non-specific binding and probed with specific antibodies targeting SNX4 and GAPDH. After incubation, protein bands were visualized using an enhanced chemiluminescence (ECL) detection system. The grayscale values of the protein bands were analyzed using ImageJ software.
Statistical analyses
All of the statistical analyses were performed by R (V 3.6.3), and the R package ggplot2 was utilized to analyze the expression differences. The Wilcoxon rank sum test Mann-Whitney U test and Student’s t-test were conducted to establish the gene expression differences among ccRCC tissues and surrounding normal tissues. Log-rank and Kaplan-Meier tests were performed to picture Kaplan-Meier survival curves by using the survminer package. Correlation analysis was conducted using the Pearson correlation and Spearman tests. All the experiments in this paper were conducted repeatedly three times, and the final experimental data were expressed as mean ± standard deviation. Unpaired t-test and one-way analysis of variance (ANOVA) analysis were applied to assess the differences between experiment and control groups. P-value lower than 0.05 was considered significant.
| Results | ▴Top |
Expression pattern of SNX4 in pan-cancer perspective
Expression pattern of SNX4 was evaluated among 33 cancer types based on the TCGA database. As revealed in Figure 1, SNX4 expression was significantly upregulated in 22 out of 33 cancer types and downregulated in five cancer types compared with normal tissues.
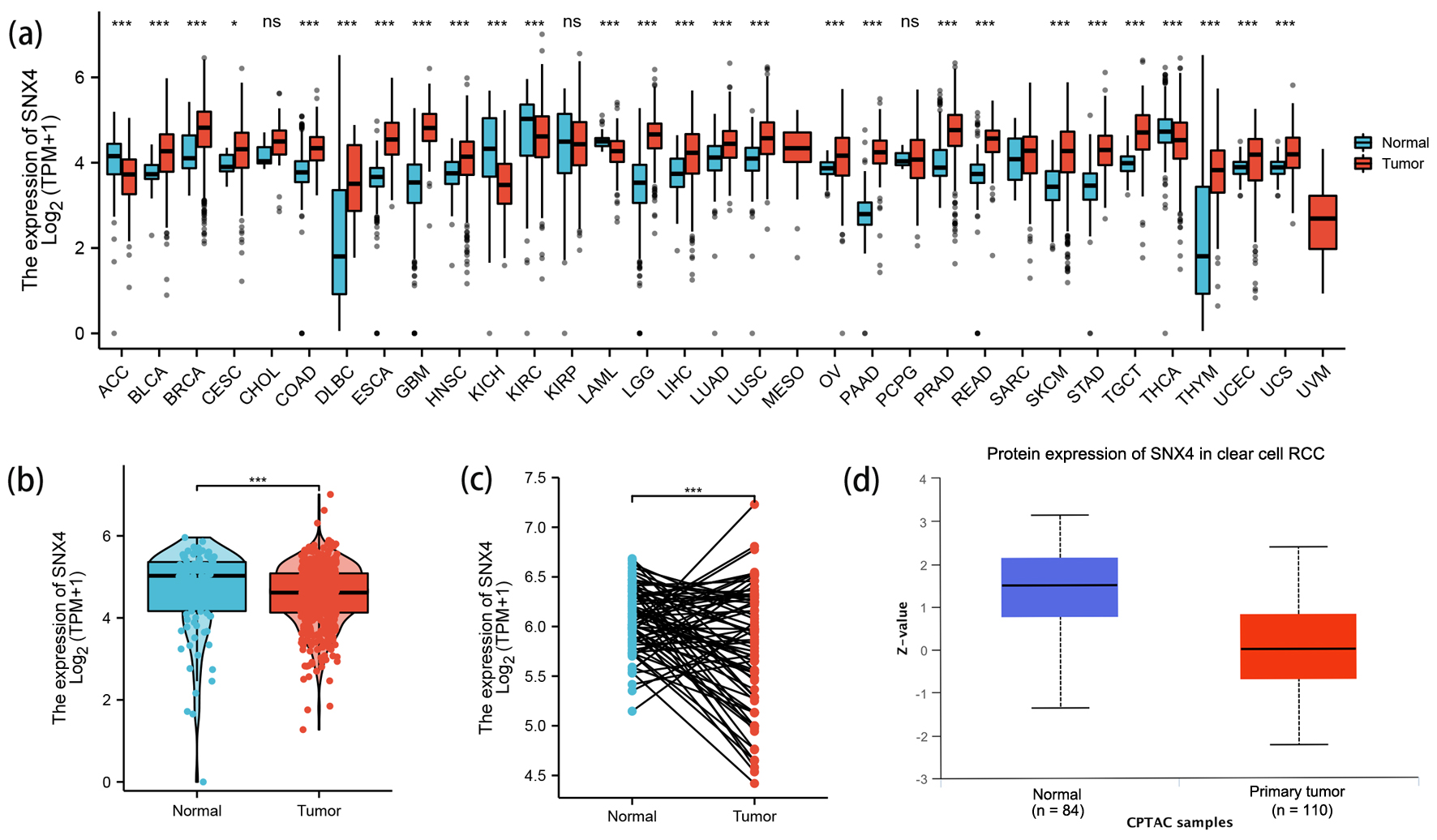 Click for large image | Figure 1. The expression levels of SNX4. (a) The expression levels of SNX4 in pan-cancer from TCGA database. (b, c) The expression levels of SNX4 in ccRCC and normal renal tissues from TCGA database. (d) The protein expression levels of SNX4 in ccRCC and normal renal tissues in CPTAC. *P < 0.05, **P < 0.01, ***P < 0.001. ns: no significance. |
Downregulated mRNA and protein expression of SNX4 in ccRCC patients
In order to determine differences of SNX4 expression in ccRCC and normal tissues, we analyzed the data on mRNA SNX4 expression levels found in TCGA and GTEx. Figure 1b shows the unpaired differential expression analysis indicating that the mRNA expression level of SNX4 in ccRCC (n = 531) was significantly lower than those in normal tissues (n = 100) (4.708 ± 1.033 vs. 4.549 ± 0.727, P < 0.001).
Next, we acquired the paired data analyses between ccRCC tissues and surrounding healthy tissues illustrating the downregulation of SNX4 in ccRCC (n = 72) (Fig. 1c, 6.083 ± 0.334 vs. 5.776 ± 0.625, P < 0.001).
We continued to investigate the protein level of SNX4 on CPTAC via UALCAN and found significantly lower expression of SNX4 in ccRCC samples (n = 110) than those in normal samples (n = 84) (Fig. 1d). Immunohistochemical staining images for SNX4 proteins in ccRCC as well as normal kidney tissues were obtained from the HPA database and shown respectively in Figure 2a-d.
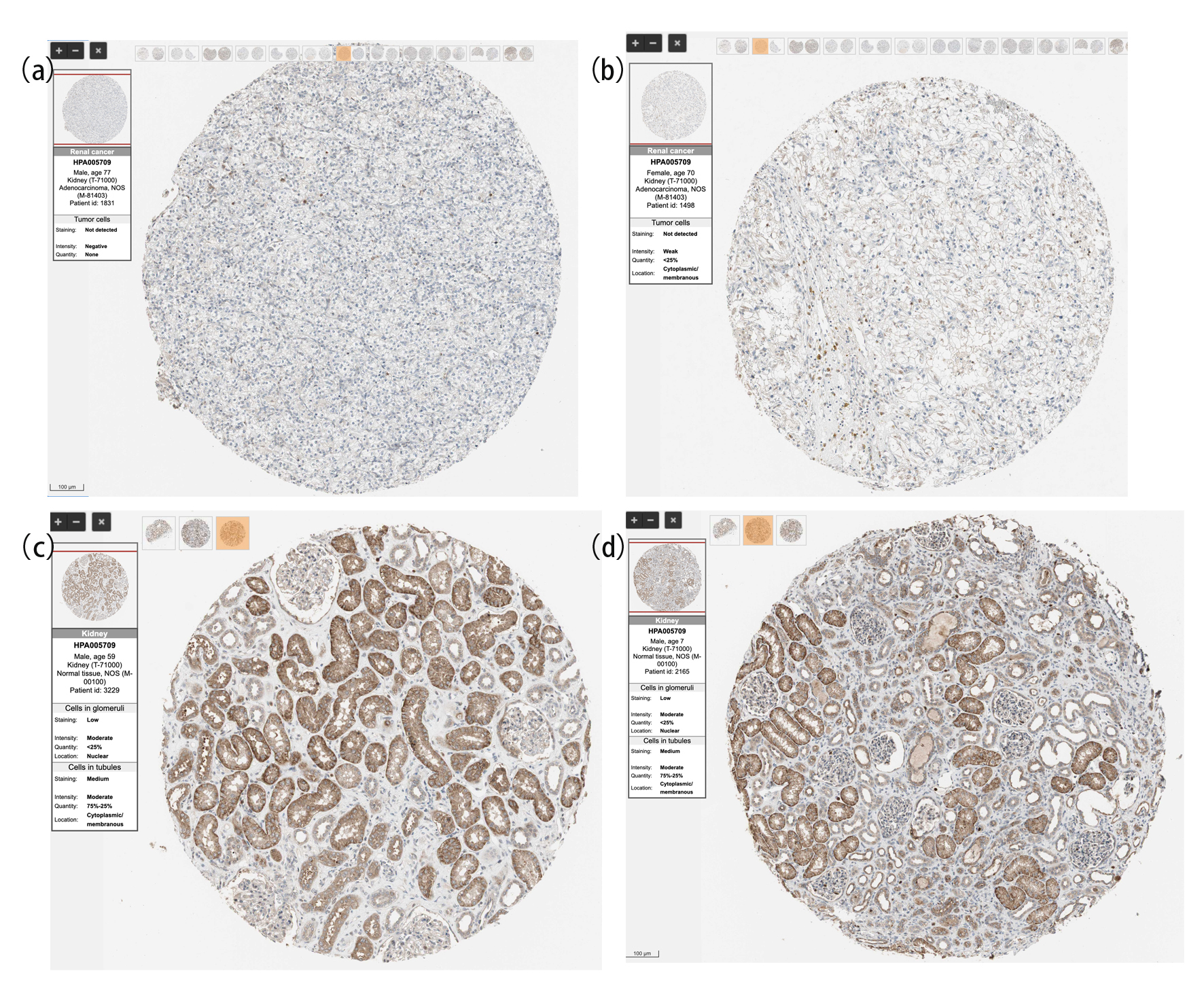 Click for large image | Figure 2. The protein expression of SNX4 in ccRCC and normal renal tissues based on the Human Protein Atlas. (a, b) The protein levels of SNX4 in normal kidney tissues from the Human Protein Atlas database. (c, d) The protein levels of SNX4 in kidney clear cell carcinoma based on the Human Protein Atlas. |
Relationships between SNX4 mRNA levels and clinical pathological features of ccRCC patients
To determine relationships between SNX4 mRNA levels and clinical pathological features of ccRCC patients, Dunn’s test and the Kruskal-Walli’s test were used.
As shown in Figure 3e, there was statistically significant reduction of SNX4 expression in patients with higher histologic grade (P < 0.1). Figure 3f demonstrates lower expression in male patients than that in female (P < 0.001). No significant difference of SNX4 expression level was observed in other clinical characteristics, including T stage (Fig. 3a), N stage (Fig. 3b), M stage (Fig. 3c), pathologic stage (Fig. 3d), age (Fig. 3g) or primary therapy outcome (Fig. 3h).
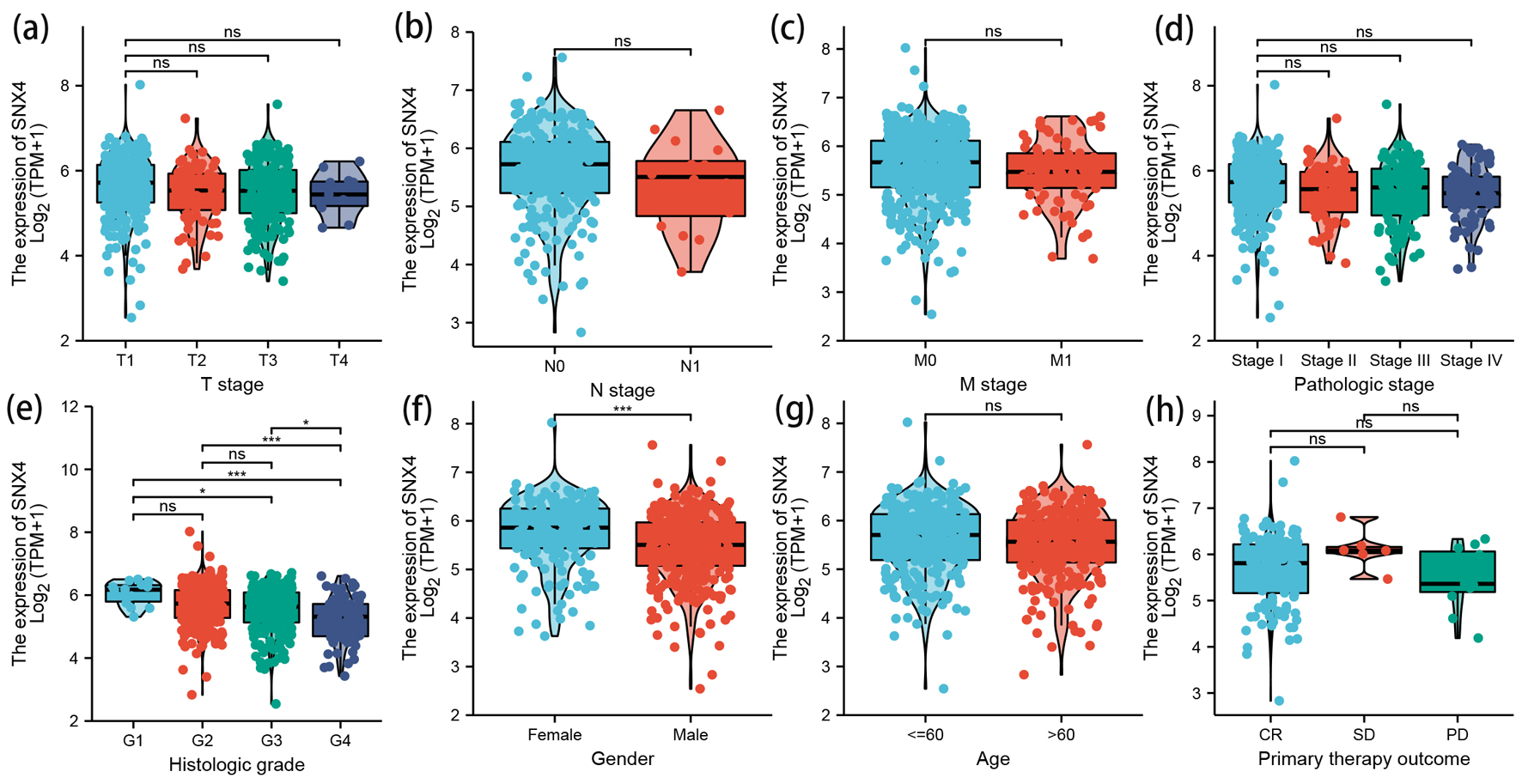 Click for large image | Figure 3. The correlation between SNX4 expression and clinical characteristics. Lower expression levels of SNX4 were observed in patients with high histologic grade (e) and male patients (f). No significant correlation was detected between the expression levels of SNX4 and T stage (a), N stage (b), M stage (c), pathologic stage (d), age (g), or primary therapy outcome (h). *P < 0.05, **P < 0.01, ***P < 0.001. ns: no significance. |
Low mRNA expression of SNX4 is correlated with poor prognosis
Kaplan-Meier analysis was conducted in TCGA KIRC cohort. Based on the SNX4 expression status, ccRCC patients were divided into high SNX4 expression group (n = 258) and low SNX4 expression group (n = 258). The cut-off value was set to be the median SNX4 mRNA level. Kaplan-Meier curves revealed that lower mRNA expression level of SNX4 was correlated with poorer OS (hazard ratio (HR) = 0.49, P < 0.001; Fig. 4a), shorter disease-specific survival (DSS) (HR = 0.36, P < 0.001; Fig. 4b) and worse progression-free interval (PFI) (HR = 0.56, P < 0.001; Fig. 4c). We further plotted the Kaplan-Meier survival curves based on distinct histologic grade (Fig. 4f-i) and gender (Fig. 4d, e). It showed that lower SNX4 expression was related to the poorer OS among G3 patients (HR = 0.41, P < 0.001; Fig. 4h) and female patients (HR = 0.31, P < 0.001; Fig. 4e).
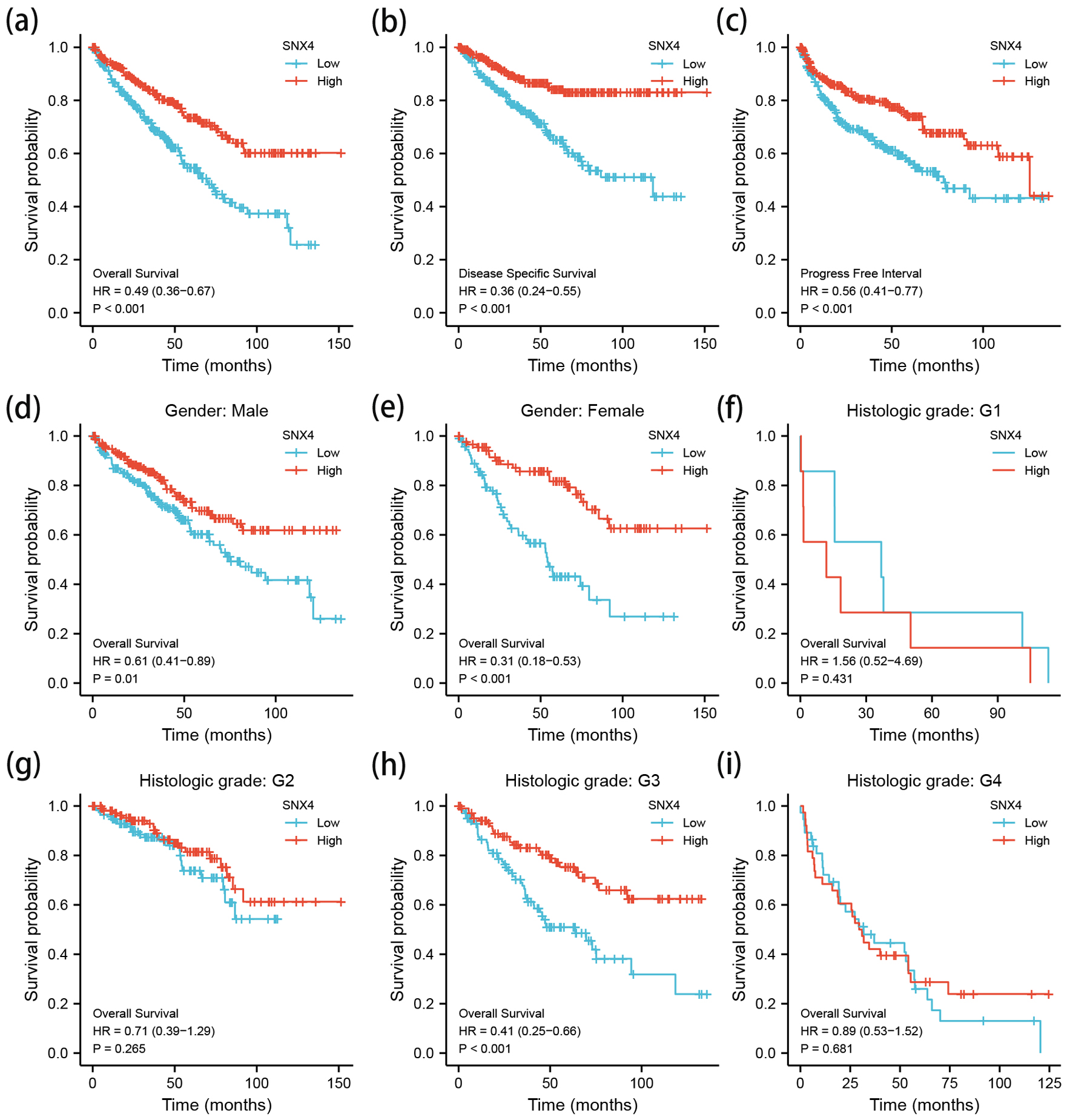 Click for large image | Figure 4. The Kaplan-Meier survival curve of ccRCC stratified according to SNX4 expression levels indicated that ccRCC patients with low SNX4 mRNA expression had a shorter OS, DSS and PFI than those with high level of SNX4 (a-c). Besides, a subgroup analysis was performed on different gender and histologic grade, respectively (d-i). OS: overall survival; DSS: disease-specific survival; PFI: progression-free interval. |
Correlated significant genes with SNX4
The correlated significant genes of SNX4 were explored using the LinkedOmics database. The first 50 correlated genes were visualized in the heatmap plot (Fig. 5a) and volcano plot (Fig. 5b). We found that SNX4 presented the strongest negative correlation with LOC729234, RIN1 and GGN while positive correlation with EHHADH, RABL3 and PLS1.
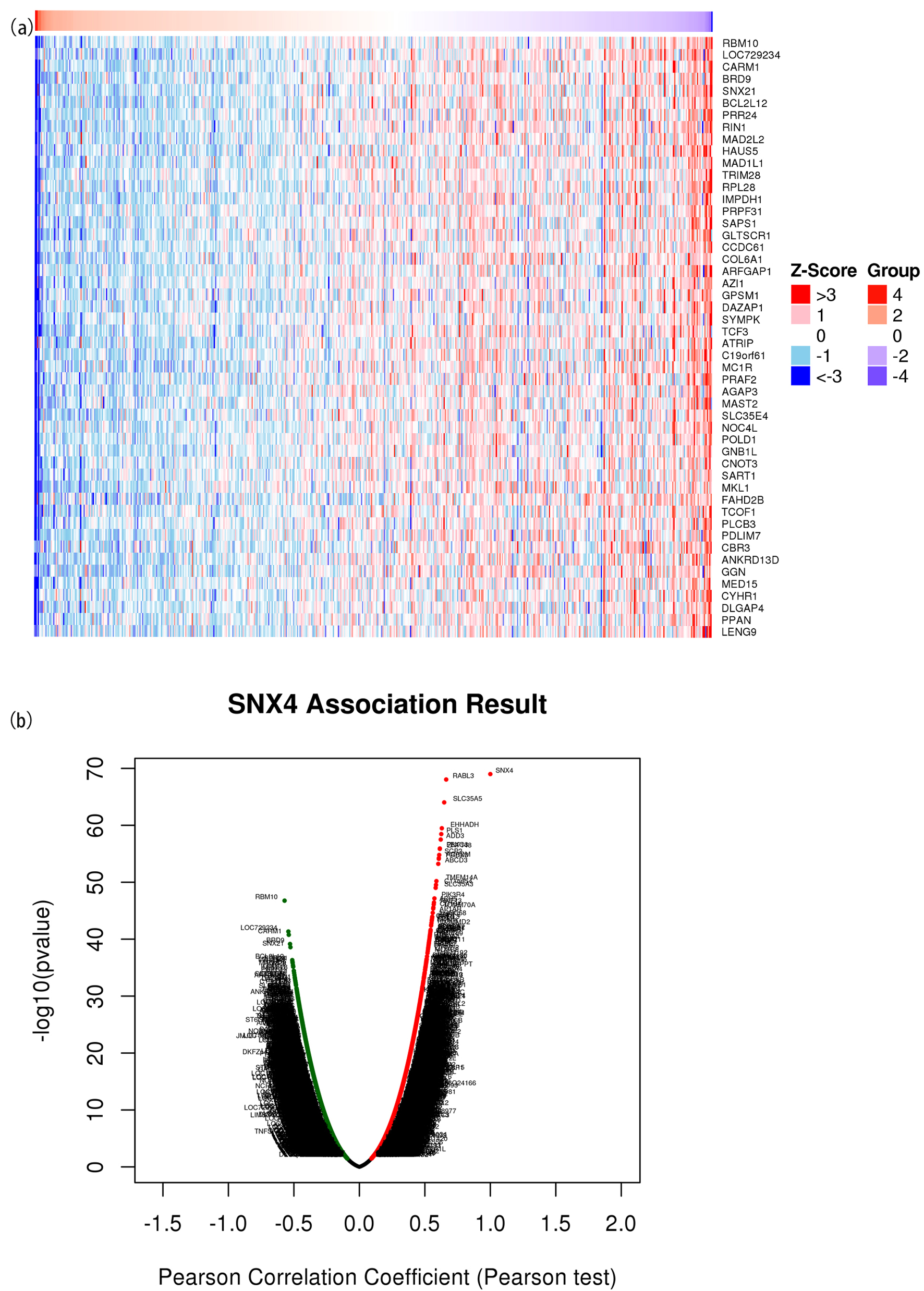 Click for large image | Figure 5. The correlated significant genes of SNX4 were visualized in the heatmap plot (a) and volcano plot (b). (a) Heatmap of the top 50 significant differentially expressed genes between the high and low SNX4 expression groups. Blue and red dots represent downregulated and upregulated genes, respectively. (b) Volcano plot of differentially expressed genes between the high and low SNX4 expression groups. |
Univariable and multivariable Cox regression
Univariate and multivariate Cox regression analyses were conducted to discern independent factors significantly associated with ccRCC OS, considering SNX4 expression and other clinical parameters (age, gender, T stage, N stage, M stage, pathologic stage, histologic grade, race, laterality and primary therapy outcome). Univariate Cox regression analysis revealed significant associations of T stage (P < 0.001), N stage (P < 0.001), M stage (P < 0.001), pathologic stage (P < 0.001), histologic grade (P < 0.001), age (P < 0.001) and SNX4 expression (P < 0.001) with OS. Multivariate Cox regression analysis demonstrated that no parameters remained independent prognostic factors for ccRCC patients (Table 1).
 Click to view | Table 1. Univariable and Multivariable Cox Regression |
Construction and verification of nomogram based on SNX4 expression
Employing multivariable Cox regression analysis, we systematically identified clinical factors influencing the prognosis of ccRCC patients. Subsequently, we devised a nomogram (depicting age, T stage, N stage, M stage, pathologic stage, histologic grade and SNX4 expression; Fig. 6a) to assist clinicians in prognostic predictions. The nomogram featured line segments, each representing a score ranging from 0 to 100, with the length of these segments signifying the impact of individual clinical factors on OS. The cumulative score, derived by summing up individual scores, enabled the determination of expected survival rates at the first, third and fifth years. Furthermore, the calibration curve underscored the favorable concordance between predicted and actual outcomes (Fig. 6b). In summary, the constructed nomogram successfully provides a robust model for accurate prognosis prediction in ccRCC patients.
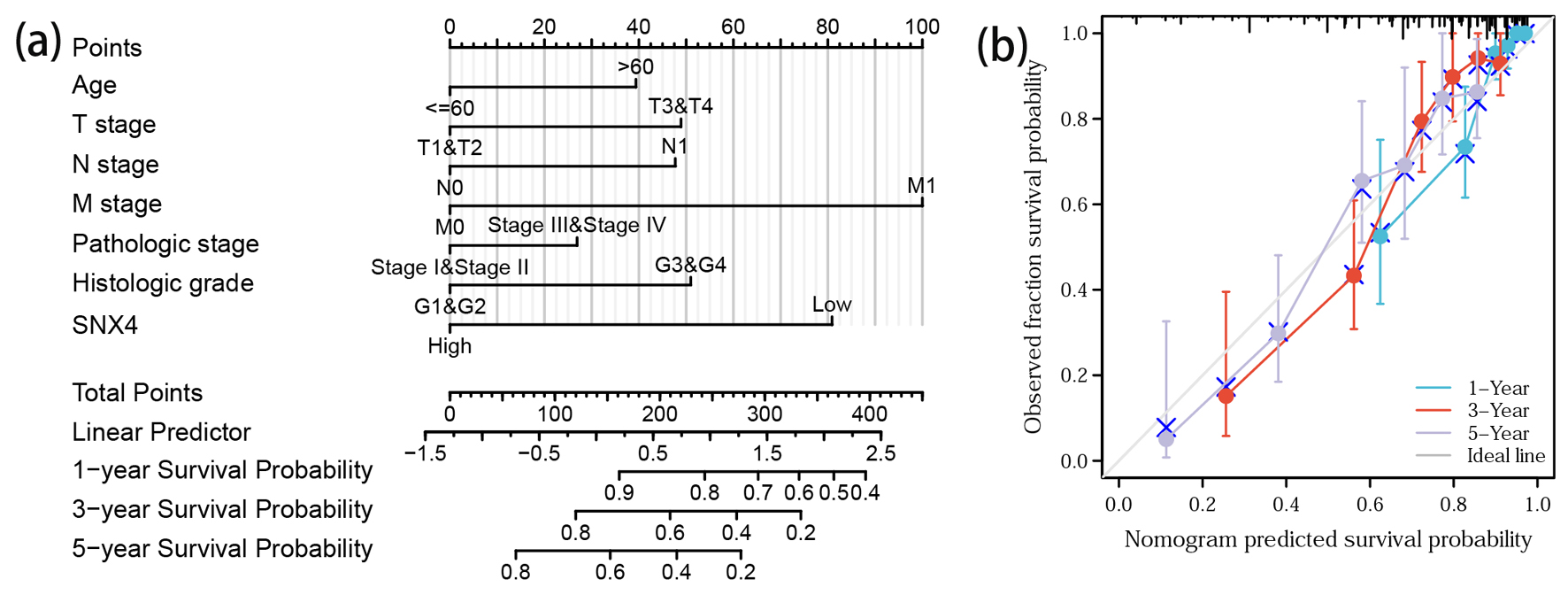 Click for large image | Figure 6. Prediction model nomogram and calibration curve. (a) Nomogram for predicting the probability of 1-, 3-, and 5-year OS for ccRCC patients. (b) Calibration plot of the nomogram for predicting the survival probability. |
Correlation analysis of SNX4 expression and immune cell infiltration in ccRCC
The TIMER database was utilized to estimate the underlying relationship between SNX4 expression level and the six diverse types of tumor-infiltrating immune cells. The results are shown in Figure 7a, suggesting that the expression level of SNX4 had positive correlation with infiltrating levels of B cells (r = 0.241, P = 1.65 × 10-7), CD8+ T cells (r = 0.188, P = 7.60 × 10-5), CD4+ T cells (r = 0.147, P = 1.55 × 10-3), macrophages (r = 0.297, P = 1.48 × 10-10), neutrophils (r = 0.269, P = 4.95 × 10-9), and dendritic cells (r = 0.203, P = 1.24 × 10-5) in ccRCC, although no significant association with tumor purity. P < 0.05 was considered as the difference of significance.
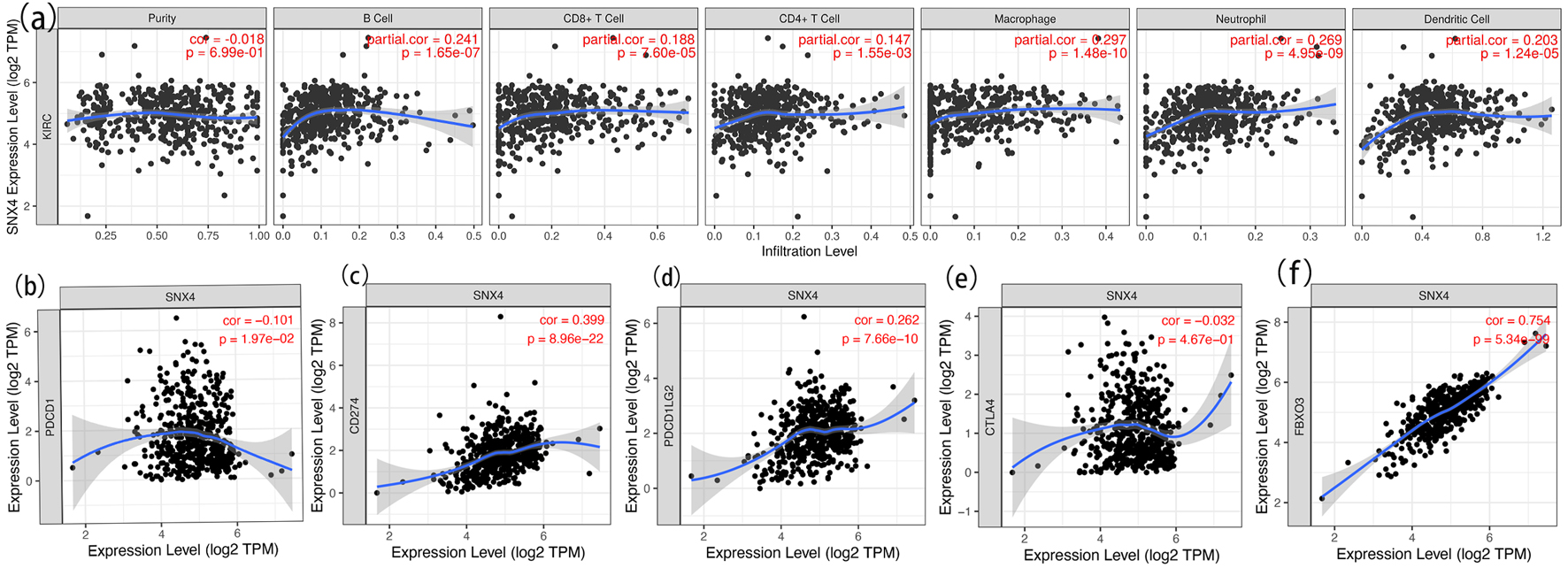 Click for large image | Figure 7. Correlation analysis of SNX4 expression and immune cell infiltration in ccRCC (a) and relationship between SNX4 and immune checkpoints of PD-1 (b), CD274 (c), PD-L1 (d), CTLA4 (e) and FBXO3 (f) in ccRCC. |
Further investigations were conducted to explore the possible role of SNX4 in tumor immune in ccRCC. We used the GEPIA database to determine the association between SNX4 and corresponding biomarkers of immune cells. As listed in Table 2, SNX4 was significantly positively correlated with CD4+ T cell biomarkers (CD4), part of M1 macrophage biomarkers (NOS2 and IRF5), M2 macrophage biomarkers (CD163, VSIG4, and MS4A4A), part of neutrophil biomarkers (CEACAM8 and ITGAM), and dendritic cell biomarkers (HLA-DPB1, HLA-DRA, HLA-DPA1, CD1C and NRP1). There was no significant association between SNX4 expression and B cell or CD8+ T cell biomarkers. These results revealed that SNX4 may regulate macrophage polarization in ccRCC.
Click to view | Table 2. Association Between SNX4 and Corresponding Biomarkers of Immune Cells |
Relationship between SNX4 and immune checkpoints in ccRCC
Considering SNX4 as the potential tumor suppressor gene in ccRCC, the relationship of SNX4 expression and immune checkpoint genes, involving PD-1, CD274, PD-L1, CTLA4 and FBXO3, was assessed and exhibited respectively in Figure 7b-f. PD1 and PD-L1 are important immune checkpoints which are responsible for tumor immune escape. As suggested in Figure 7d, SNX4 expression was significantly positively correlated with PD-L1 in ccRCC, while Figure 7b displays nonsignificant correlation between PD-1 and SNX4 expression. These results demonstrate that tumor immune escape might be involved in SNX4-mediated carcinogenesis of ccRCC.
Prediction and analysis of upstream miRNAs of SNX4
It was generally accepted that mRNA-miRNA-lncRNA ceRNA hypothesis was a new regulatory mechanism between ncRNA and mRNA. To construct the ceRNA network, the underlying upstream miRNAs obtained from four databases were intersected using Venn analysis to ascertain six miRNAs with closer possibility and visualized in Venn diagram webtool [37] (Fig. 8a). The miRNAs and SNX4 interactions were performed and listed in Figure 8b-e, suggesting that SNX4 was significantly negatively correlated with miR-34c-5p, miR-221-3p, miR-218-5p and miR-222-3p in ccRCC. We further explored the expression and prognostic influence of the four miRNAs as demonstrated in Figure 8f-i. MiR-221-3p and miR-222-3p were significantly upregulated in ccRCC samples and their overexpression was correlated with worse OS. These findings supported that miR-221-3p and miR-222-3p might be the possible regulatory upstream miRNAs of SNX4 in ccRCC.
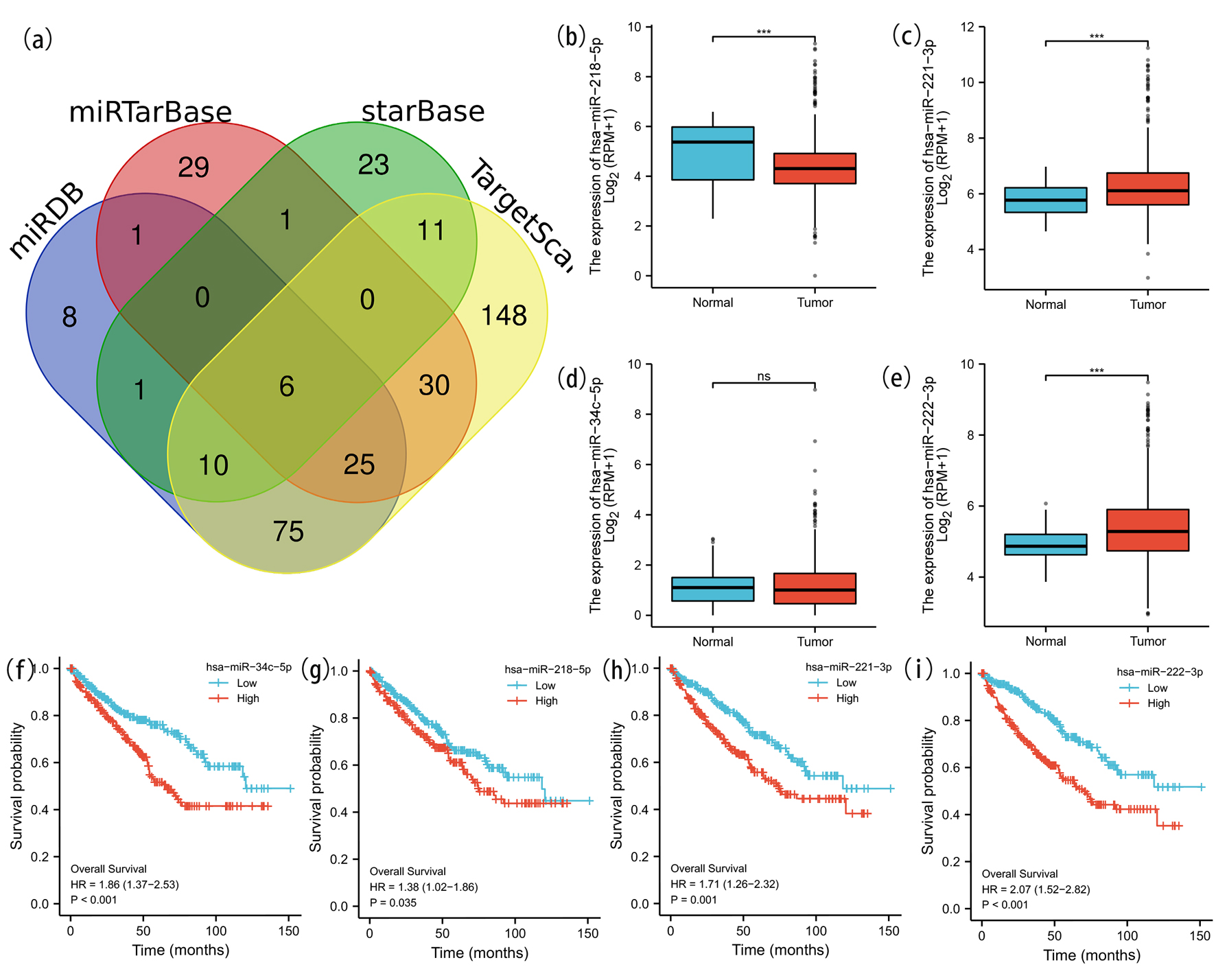 Click for large image | Figure 8. Prediction and analysis of ceRNA network. (a) Venn diagrams of underlying miRNA were drawn to predict the mutual target miRNA. The expression levels of miR-218-5p (b), miR-221-3p (c) and miR-222-3p (e) were significantly different in ccRCC and normal renal tissue. No significant difference of the expression level of miR-34c-5p (d) was detected in ccRCC and normal tissue. We further explored the expression and prognostic influence of the four miRNAs (f-i). |
Prediction and analysis of upstream lncRNAs of miR-221-3p/miR-222-3p
By searching in starBase database, we acquired basically the same possible upstream lncRNAs of miR-221-3p/miR-222-3p. The expression level of associated lncRNAs was acquired through GEPIA. Log2FoldChange and P value were screened to select lncRNAs with significantly downregulated expression. Based on the ceRNA hypothesis, lncRNA could competitively bind to shared miRNAs and increase mRNA expression level. Thus, underlying lncRNAs were negatively correlated with miRNA and positively correlated with mRNA. The correlation analysis between lncRNAs and miR-221-3p/miR-222-3p or lncRNAs and SNX4 in ccRCC was assessed by starBase database and the final result was listed in Table 3. Taken together, DHRS4-AS1 was most likely the shared upstream lncRNA of miR-221-3p/miR-222-3p in ccRCC.
Click to view | Table 3. Prediction and Analysis of ceRNA Network |
Validation of the expression level of SNX4 in ccRCC
The qRT-PCR analysis revealed a significant downregulation of SNX4 expression in ccRCC samples compared to the corresponding adjacent normal tissues (Fig. 9a, b). Raw data are provided in the Supplementary Material 1 (www.wjon.org). Protein expression levels of SNX4 were checked by WB in both ccRCC tissues and adjacent non-ccRCC tissues. A significant reduction in SNX4 expression was observed in the cancerous tissues compared to the corresponding nccRCC tissues (Fig. 9d) as WB gray scale analysis shown in Figure 9c.
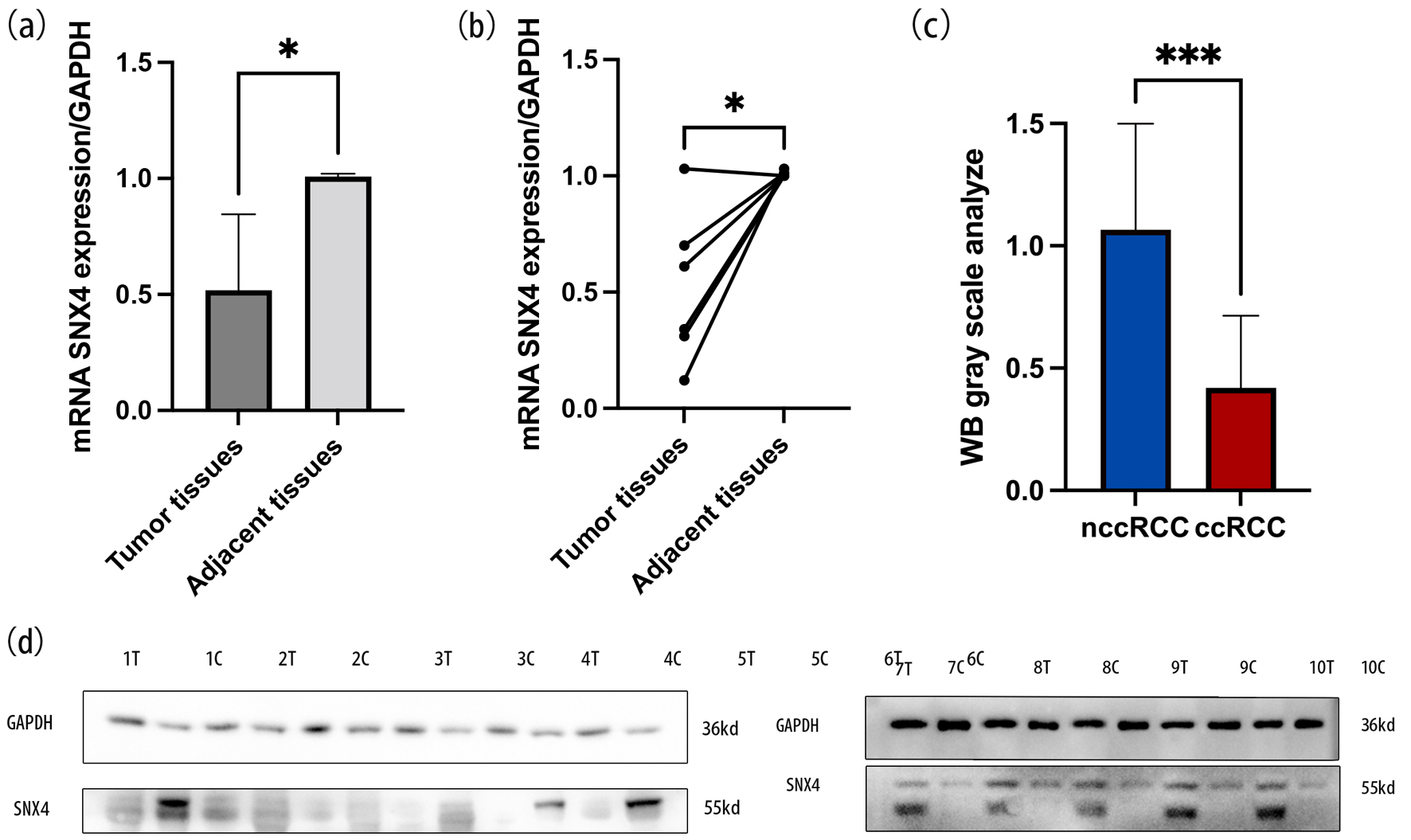 Click for large image | Figure 9. The mRNA expression levels of SNX4 in ccRCC tissues and normal tissues (a, b). WB gray scale analysis indicated the protein expression levels of SNX4 in ccRCC tissues and normal tissues (c). Western blot indicated that the protein expression levels of SNX4 were downregulated in ccRCC compared with normal renal tissue (d). *P < 0.05, ***P < 0.001. |
| Discussion | ▴Top |
SNX4, also known as ATG24B (autophagy protein 24B), is an important regulator concerning the retrieval of cargo from degeneration into a recycling pathway [17]. Our research was the first effort to explore the correlation between SNX4 and ccRCC via bioinformatics technology. It was generally reported that SNX4 was highly expressed in normal kidney as compared to other tissues. According to our research, SNX4 acted as a tumor suppressor gene in ccRCC. Both mRNA and protein expression of SNX4 were downregulated in ccRCC samples. The expression levels of SNX4 were further identified by qRT-PCR and WB in our clinical samples. And its low expression level was significantly correlated to high histologic grade and resulted in different outcomes on different genders. As Kaplan-Meier curves suggested, short OS and DFS were identified with the depressed mRNA expression of SNX4, which enables SNX4 potentially to act as a predictive biomarker related to a favorable prognosis in ccRCC. We further identified that SNX4 exerted a distinct function in immune infiltration in ccRCC. The expression level of SNX4 was positively correlated with the immune checkpoint expression level, including PDCD1LG2 and FBXO3, and we also listed its affiliation with the immune cell biomarker in Table 2. Aberrant miRNA expression is implicated in various tumorous biological processes. Based on the theory of ceRNA network, we screened the most closely related miRNA using Tarbase database and then explored the possible upstream lncRNA, which was listed in Table 3. Finally, SNX4/miR-221/222-3p/DHRS4-AS1 regulatory axis was successfully constructed in ccRCC and we simultaneously explored the expression level and prognostic value of genes involved in the pathway.
The lysosomal degradation pathway of autophagy plays a fundamental role in cellular, tissue and organismal homeostasis and, in part, is mediated by the repression and activation of ATG genes [38]. Experimentally, ATG proteins interactions are known to be important for autophagosome formation [38]. Autophagy can promote or inhibit apoptosis under different cellular contexts in response to stimuli due to the degradation of different pro- or anti-apoptotic regulators by autophagy [39]. In the aging model Podospora anserina, ablation of PaATG24 leads to a reduced growth rate, infertility, and to a pronounced lifespan reduction [40]. Several experiments have identified that SNX4 was involved in autophagy initiation and mediated ATG9A trafficking, which drove expansion of nascent autophagosome membranes, from endolysosomes to early endosomes [15, 41, 42]. ATG20 forms a heterodimer complex with SNX4 via its BAR domain and the complex tubulates liposomes influencing the overall shape of the phagophore membrane during autophagy [43, 44]. Efficient transfer of Ssn2/Med13, which act as repressor of ATG expression, from the nuclear periphery to phagophores depends on the sorting nexin heterodimer Snx4-Atg20, which binds to Atg17, and relocates to the perinucleus in response to nitrogen starvation [16]. It suggests that SNX4 exerts its influence on autophagy by interacting with multiple ATG genes, indicating a possible impact on cancer development [38, 45]. However, the explicit mechanism of SNX4 in ccRCC remains largely unclear and needs to be further explored.
Ferroptosis, a novel form of oxidative stress-induced cell death, is driven by lethal lipid peroxidation and accumulated iron [46]. Iron exerts a central role in cell viability and death, and cellular iron homeostasis is under exquisite control [47]. Many cellular processes alter the sensitivity of cells towards ferroptosis by changing cellular labile iron contents. Transferrin and its receptor work together to facilitate ferroptosis by importing iron into the cell [48]. SNX4 coordinates transporting transferrin receptor (TfnR) with long range from early endosomes to the juxtanuclear endocytic recycling compartment [49]. Suppression of SNX4 perturbs the transport and causes lysosomal degradation of TfnR. In an endocytosis assay, overexpressed SNX4 was able to inhibit TfnR endocytosis, and SNX4 colocalized with transferrin-containing vesicles at lower levels of expression, suggesting that SNX4 may be part of the endocytic machinery or, alternatively, that SNX4 is likely to relate to key elements of endocytosis and sequester them when overexpressed [50]. As cancer cell is characterized by more active metabolism and higher reactive oxygen species (ROS) load, it may have higher tendency to undergo ferroptosis. Adding to this notion, cancer cell often demands high iron supply, which enhances their susceptibility to ferroptosis [51, 52]. Based on the above experimental results, we make a speculation that downexpression of SNX4 in ccRCC may inhibit TfnR endocytosis and block the intracellular transport of iron, which may restrain the ferroptosis process.
A key issue surrounding tumor formation includes the influence that the immune system puts on resisting or eradicating formation and progression of neoplasm. Immune surveillance system monitors cells and tissues constantly and takes charge of recognizing and eliminating the vast majority of incipient cancer cells [53]. So there is reason to believe ccRCCs that do appear have somehow managed to avoid the immunological killing, thereby evading immune eradication [54]. ATG proteins mediate inflammatory and immune signaling transport simultaneously through autophagy-dependent mechanisms and autophagy-independent mechanisms, which generally implicate ATG protein crosstalk with immune signaling molecules [38]. To further investigate the role that SNX4 plays in immune escape, we assessed the correlations between its expression and immune infiltration level in ccRCC. We propose that increased activation of SNX4 expression arouses the accumulation activation of a range of distinct immune cell types and can markedly rearrange the tumor immune microenvironment (TME) through various mechanisms, including cytokine secretion and metabolic derangements. ICB therapies unleash breaks in the immune system and generate durable responses [55]. However, patients present unwantedly low response rates to ICB therapy owing to the unexpected recycling and cellular abundance of PD-L1 [56]. Thus, understanding the underlying molecular mechanisms better will assist the establishment of novel strategies to treat and prevent resistance. Autophagy pathway plays a key role in survival and apoptosis, activation, differentiation, effector function and tumor trafficking of extensive immune cell subsets, and meanwhile alters tumor immune response through modulating autophagy pathway in cancer therapy. Inducing the lysosomal degradation of PD-L1 antibody and the consequent decrease in its cellular abundance can be a promising approach for reliable and durable blockade of PD-L1 to increase the ICB therapy efficiency and response rate of patients. Autophagy inhibition plus anti-PD-L1 is an attractive combination for further investigation, particularly for tumors with high levels of autophagy. SNX4, as an autophagy gene, may lead to a significant PD-L1 degradation that prevents its recycling and cellular abundance compared to anti-PD-L1 antibody, disrupting immune escape mechanism of tumor cells and enhancing T cell-mediated antitumor immunity. It may potentially provide predictive biomarkers and mechanisms to assess treatment efficacy. Based on our current research, expression level of SNX4 has an impact on the abundance and activation of immune cell, consequently converting the tumor immune environment and affect the tumor immune surveillance.
CeRNA hypothesis suggests that the ceRNA activity forms a large-scale regulatory network, which is key post-transcriptional regulators of gene expression and has wide implications in pathological conditions, such as cancer [57]. SNX4, interacting with miRNAs or lnRNAs, may serve for ceRNAs as a basic component of the ceRNA regulatory network and be pivotal in the biological properties of ccRCC. However, it still remains unelucidated how SNX4 mediates the ceRNA-ceRNA interaction network and participates in the development and growth of ccRCC. In vitro experiments revealed that microRNA-222-3p promotes tumor cell migration and invasion and inhibits apoptosis, and is correlated with an unfavorable prognosis of patients with RCC [58]. SNX4, acted as its target gene, was negatively correlated with miR-222-3p. Several researches have verified the altered expression level and clinical significance of miR-221-3p and miR-222-3p. Our study further constructed the overall ceRNA network and also evaluates the potential clinical utility of the ceRNA axis, SNX4/miR-221/222-3p/DHRS4-AS1 axis, and their possible relations with clinical prognosis. The analyses of SNX4-associated ceRNA network provided an improved outlook of the mechanism of action of ccRCC and may provide a novel direction into clinical application in ccRCC. Downregulation of DHRS4-AS1 in ccRCC was confirmed by assessing a cohort of tumor and paired non-tumor samples [59]. Low DHRS4-AS1 expression was significantly associated with advanced pathological grade and poor prognosis, which simultaneously verified our prediction [59].
We also found that our study has some limitations. First of all, we only verified the mRNA and protein expression of the target gene using qRT-PCR and WB. More clinical samples are needed to confirm its expression level and correlation with clinical characteristics. Further laboratory validation is required before any conclusions can be made about the effect of SNX4 on ccRCC. Second, we did not interrogate the molecular mechanisms by which SNX4 may exert its protective effects in ccRCC.
Conclusion
The mRNA and protein expression level of SNX4 was significantly lower in ccRCC than those in normal tissues. Lower mRNA expression level of SNX4 was correlated with poor prognosis. SNX4 may exert critical roles in the tumorigenesis, development and migration of ccRCC via various mechanisms.
| Supplementary Material | ▴Top |
Suppl 1. Expression level of SNX4.
Acknowledgments
None to declare.
Financial Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Conflict of Interest
All authors of accepted articles disclosed no any conflict of interest.
Informed Consent
Participants gave written informed consents.
Author Contributions
Yong Zhang and Yuan Shan Cui designed the research and revised the paper. Yu Meng Chai, Zhong Bao Zhou and Run Ze Liu performed the data analysis and experiments. Yu Meng Chai drafted the paper. All of the authors approved the submitted and final versions.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, Heng DY, et al. Renal cell carcinoma. Nat Rev Dis Primers. 2017;3:17009.
doi pubmed pmc - Capitanio U, Bensalah K, Bex A, Boorjian SA, Bray F, Coleman J, Gore JL, et al. Epidemiology of renal cell carcinoma. Eur Urol. 2019;75(1):74-84.
doi pubmed pmc - Ricketts CJ, De Cubas AA, Fan H, Smith CC, Lang M, Reznik E, Bowlby R, et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018;23(1):313-326.e315.
doi pubmed pmc - Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1-2):48-61.
doi pubmed pmc - Motzer RJ, Jonasch E, Michaelson MD, Nandagopal L, Gore JL, George S, Alva A, et al. NCCN Guidelines Insights: Kidney Cancer, Version 2.2020. J Natl Compr Canc Netw. 2019;17(11):1278-1285.
doi pubmed - Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, Venugopal B, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380(12):1103-1115.
doi pubmed pmc - Motzer RJ, Rini BI, McDermott DF, Aren Frontera O, Hammers HJ, Carducci MA, Salman P, et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019;20(10):1370-1385.
doi pubmed pmc - Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803-1813.
doi pubmed pmc - Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer. 2019;19(3):133-150.
doi pubmed pmc - Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171(4):934-949.e916.
doi pubmed pmc - Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43-49.
doi pubmed pmc - Choueiri TK, Kaelin WG, Jr. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat Med. 2020;26(10):1519-1530.
doi pubmed - Cullen PJ, Korswagen HC. Sorting nexins provide diversity for retromer-dependent trafficking events. Nat Cell Biol. 2011;14(1):29-37.
doi pubmed pmc - Yang L, Tan W, Yang X, You Y, Wang J, Wen G, Zhong J. Sorting nexins: A novel promising therapy target for cancerous/neoplastic diseases. J Cell Physiol. 2021;236(5):3317-3335.
doi pubmed - Anton Z, Betin VMS, Simonetti B, Traer CJ, Attar N, Cullen PJ, Lane JD. A heterodimeric SNX4—SNX7 SNX-BAR autophagy complex coordinates ATG9A trafficking for efficient autophagosome assembly. J Cell Sci. 2020;133(14):jcs246306.
doi pubmed pmc - Hanley SE, Willis SD, Cooper KF. Snx4-assisted vacuolar targeting of transcription factors defines a new autophagy pathway for controlling ATG expression. Autophagy. 2021;17(11):3547-3565.
doi pubmed pmc - Zhou C, Wu Z, Du W, Que H, Wang Y, Ouyang Q, Jian F, et al. Recycling of autophagosomal components from autolysosomes by the recycler complex. Nat Cell Biol. 2022;24(4):497-512.
doi pubmed - Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353-358.
doi pubmed pmc - https://genome-cancer.ucsc.edu/.
- http://www.ncbi.nlm.nih.gov/geo/.
- https://www.proteinatlas.org/.
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419.
doi pubmed - http://ualcan.path.uab.edu/.
- Chandrashekar DS, Karthikeyan SK, Korla PK, Patel H, Shovon AR, Athar M, Netto GJ, et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia. 2022;25:18-27.
doi pubmed pmc - Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, Varambally S. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19(8):649-658.
doi pubmed pmc - http://ualcan.path.uab.edu/analysis-prot.html.
- http://linkedomics.org/.
- Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018;46(D1):D956-D963.
doi pubmed pmc - http://timer.cistrome.org/
- Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509-W514.
doi pubmed pmc - http://gepia.cancer-pku.cn/index.html.
- Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, Vlachos IS, Tastsoglou S, Kanellos I, Papadimitriou D, et al. DIANA-TarBase v8: a decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018;46(D1):D239-D245.
doi pubmed pmc - Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48(D1):D127-D131.
doi pubmed pmc - Liu W, Wang X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019;20(1):18.
doi pubmed pmc - Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42(Database issue):D92-D97.
doi pubmed pmc - Huang HY, Lin YC, Li J, Huang KY, Shrestha S, Hong HC, Tang Y, et al. miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020;48(D1):D148-D154.
doi pubmed pmc - http://bioinformatics.psb.ugent.be/webtools/Venn/.
- Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176(1-2):11-42.
doi pubmed pmc - Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528-542.
doi pubmed pmc - Henkel V, Schurmanns L, Brunner M, Hamann A, Osiewacz HD. Role of sorting nexin PaATG24 in autophagy, aging and development of Podospora anserina. Mech Ageing Dev. 2020;186:111211.
doi pubmed - Popelka H, Damasio A, Hinshaw JE, Klionsky DJ, Ragusa MJ. Structure and function of yeast Atg20, a sorting nexin that facilitates autophagy induction. Proc Natl Acad Sci U S A. 2017;114(47):E10112-E10121.
doi pubmed pmc - Ravussin A, Brech A, Tooze SA, Stenmark H. The phosphatidylinositol 3-phosphate-binding protein SNX4 controls ATG9A recycling and autophagy. J Cell Sci. 2021;134(3):jcs250670.
doi pubmed pmc - Popelka H, Klionsky DJ, Ragusa MJ. An atypical BAR domain protein in autophagy. Autophagy. 2018;14(7):1155-1156.
doi pubmed pmc - Ma M, Kumar S, Purushothaman L, Babst M, Ungermann C, Chi RJ, Burd CG. Lipid trafficking by yeast Snx4 family SNX-BAR proteins promotes autophagy and vacuole membrane fusion. Mol Biol Cell. 2018;29(18):2190-2200.
doi pubmed pmc - Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30(17):1913-1930.
doi pubmed pmc - Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266-282.
doi pubmed pmc - Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021-1032.
doi pubmed pmc - Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, 3rd, Kang R, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425-1428.
doi pubmed pmc - Traer CJ, Rutherford AC, Palmer KJ, Wassmer T, Oakley J, Attar N, Carlton JG, et al. SNX4 coordinates endosomal sorting of TfnR with dynein-mediated transport into the endocytic recycling compartment. Nat Cell Biol. 2007;9(12):1370-1380.
doi pubmed - Leprince C, Le Scolan E, Meunier B, Fraisier V, Brandon N, De Gunzburg J, Camonis J. Sorting nexin 4 and amphiphysin 2, a new partnership between endocytosis and intracellular trafficking. J Cell Sci. 2003;116(Pt 10):1937-1948.
doi pubmed - Manz DH, Blanchette NL, Paul BT, Torti FM, Torti SV. Iron and cancer: recent insights. Ann N Y Acad Sci. 2016;1368(1):149-161.
doi pubmed pmc - Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell. 2018;33(5):890-904.e895.
doi pubmed pmc - Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646-674.
doi pubmed - Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31(6):220-227.
doi pubmed pmc - Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer. 2020;20(9):516-531.
doi pubmed pmc - Yang S, Shim MK, Song S, Cho H, Choi J, Jeon SI, Kim WJ, et al. Liposome-mediated PD-L1 multivalent binding promotes the lysosomal degradation of PD-L1 for T cell-mediated antitumor immunity. Biomaterials. 2022;290:121841.
doi pubmed - Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344-352.
doi pubmed pmc - Zhao L, Quan J, Li Z, Pan X, Wang J, Xu J, Xu W, et al. MicroRNA-222-3p promotes tumor cell migration and invasion and inhibits apoptosis, and is correlated with an unfavorable prognosis of patients with renal cell carcinoma. Int J Mol Med. 2019;43(1):525-534.
doi pubmed - Wang C, Wang G, Zhang Z, Wang Z, Ren M, Wang X, Li H, et al. The downregulated long noncoding RNA DHRS4-AS1 is protumoral and associated with the prognosis of clear cell renal cell carcinoma. Onco Targets Ther. 2018;11:5631-5646.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.