

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 15, Number 5, October 2024, pages 744-757

Targeting SNAI1-Mediated Colorectal Cancer Chemoresistance and Stemness by Sphingosine Kinase 2 Inhibition
Harinarayanan Janakiramana, Zachary Gaoa, Yun Zhub, Jiangling Donga, Scott A. Beckerc, Alhaji Jannehd, Besim Ogretmend, E. Ramsay Campa, e, f, g ![]()
aMichael E. DeBakey Department of Surgery, Baylor College of Medicine, Houston, TX 77030, USA
bMD Anderson Cancer Center, Houston, TX 77030, USA
cMolecular and Systems Pharmacology, Emory University, Atlanta, GA 30322, USA
dDepartment of Biochemistry and Molecular Biology, Medical University of South Carolina, Charleston, SC 29425, USA
eDan L. Duncan Comprehensive Cancer Center, Houston, TX 77030, USA
fMichael E. DeBakey VA Medical Center, Houston, TX 77030, USA
gCorresponding Author: E. Ramsay Camp, Division of Surgical Oncology, Michael E. DeBakey Department of Surgery, Baylor College of Medicine, Houston, TX 77030, USA
Manuscript submitted May 3, 2024, accepted July 30, 2024, published online September 16, 2024
Short title: SPHK2 Inhibition in SNAI1-Mediated CRC Resistance
doi: https://doi.org/10.14740/wjon1890
| Abstract | ▴Top |
Background: Epithelial-to-mesenchymal transition (EMT), cancer stem cells (CSCs), and colorectal cancer (CRC) therapy resistance are closely associated. Prior reports have demonstrated that sphingosine-1-phosphate (S1P) supports stem cells and maintains the CSC phenotype. We hypothesized that the EMT inducer SNAI1 drives S1P signaling to amplify CSC self-renewal capacity and chemoresistance.
Methods: CRC cell lines with or without ectopic expression of SNAI1 were used to study the role of S1P signaling as mediators of cancer stemness and 5-fluorouracil (5FU) chemoresistance. The therapeutic ability of sphingosine kinase 2 (SPHK2) was assessed using siRNA and ABC294640, a SPHK2 inhibitor. CSCs were isolated from patient-derived xenografts (PDXs) and assessed for SPHK2 and SNAI1 expression.
Results: Ectopic SNAI1 expressing cell lines demonstrated elevated SPHK2 expression and increased SPHK2 promoter activity. SPHK2 inhibition with siRNA or ABC294640 ablated in vitro self-renewal and sensitized cells to 5FU. CSCs isolated from CRC PDXs express increased SPHK2 relative to the non-CSC population. Combination ABC294640/5FU therapy significantly inhibited tumor growth in mice and enhanced 5FU response in therapy-resistant CRC patient-derived tumor organoids (PDTOs).
Conclusions: SNAI1/SPHK2 signaling mediates cancer stemness and 5FU resistance, implicating S1P as a therapeutic target for CRC. The S1P inhibitor ABC294640 holds potential as a therapeutic agent to target CSCs in therapy refractory CRC.
Keywords: Colorectal cancer; Epithelial-to-mesenchymal transition; Sphingosine; Chemotherapy resistance; Cancer stem cells
| Introduction | ▴Top |
Despite decades of research leading to advancements in the understanding of colorectal cancer (CRC) pathophysiology, CRC is still among the most lethal malignancies in the world [1] and remains the second leading cause of cancer-related deaths in the United States [2]. Although major advances have occurred in the number of effective chemotherapy options for CRC, 5-fluorouracil (5FU) remains the backbone of most combinations [3, 4]. Unfortunately, chemoresistance remains a central issue that shortens the survival of many patients. Therefore, strategies to overcome CRC chemoresistance are required to improve CRC outcomes.
A growing body of evidence across a variety of cancers including CRC supports the central role of cancer stem cells (CSCs) in tumor dissemination and therapy resistance [5-8]. Thus, CSCs represent an attractive therapy target. Epithelial-to-mesenchymal transition (EMT) transcriptional factors such as SNAI1 (also known as SNAIL) are well-described drivers of the CSC phenotype [9-11]. In CRC, SNAI1 is highly expressed in tumor-initiating cells isolated from patient-derived xenografts (PDXs) as well as from CRC-derived colonospheres [12, 13]. Furthermore, ectopic SNAI1 expression conferred colon cancer cells with stem-like phenotypes and resistance to conventional therapy, including radiation [10, 13]. Intriguingly, EMT and SNAI1 expression can induce the expression of the enzyme thymidylate synthase (TS), which is associated with resistance to 5FU in multiple cancers, including CRC [14]. Conversely, silencing TS expression in CRC cells was found to reverse EMT and reduce the protein expression level of SNAI1 [15]. Unfortunately, therapeutic strategies disrupting EMT signaling and SNAI1 expression remain elusive, and clinically translatable strategies have not been well established.
The balance among bioactive sphingolipids, including pro-apoptotic ceramides and anti-apoptotic sphingosine-1-phosphate (S1P), influences diverse programs related to angiogenesis, tumor cell survival, proliferation, and metastasis [16-19]. Sphingosine kinases 1 and 2 (SPHK1 and SPHK2) have different kinetic properties and differ in their developmental and tissue expression [20]. For example, SPHK1 is primarily active in the cytoplasm and is involved in active remodeling and maintenance of plasma membrane homeostasis [21]. Conversely, SPHK2 primarily functions as a regulator of gene expression in the nucleus, in addition to demonstrating the ability to stabilize telomerase via S1P [22, 23]. Considering the anti-apoptotic properties of S1P, numerous inhibitors have been developed targeting this pathway [24-28]. Of particular interest to CRC is ABC294640 (Opaganib, Red Hill Biopharma), an orally available small molecule SPHK2 inhibitor that demonstrated antitumor activity in a phase 1 clinical trial for solid tumors, as well as in multiple pre-clinical cancer model systems [29-33]. ABC294640 has previously been shown to significantly reduce HT29 colon cancer cell line xenograft growth and suppress colitis-induced tumor formation [34, 35]. Multiple reports demonstrate that treatment with ABC294640 inhibited the central stem cell transcriptional factor, c-MYC, suggesting it may effectively target CSCs and may serve as an agent to overcome CRC therapy resistance [32, 36-38]. Thus, we hypothesized that SNAI1 drives dysfunctional sphingolipid metabolism by activating S1P signaling pathways to amplify CSC self-renewal capacity and decrease chemotherapy sensitivity. Conversely, we hypothesize that SPHK2 inhibition will serve as a strategy to overcome SNAI1-mediated chemoresistance. Considering the safety of ABC294640 in a phase 1 clinical trial for the treatment of patients with solid tumors, this line of investigation has immediate clinical relevance and potential to improve therapy options for advanced CRC.
| Materials and Methods | ▴Top |
Data mining
To determine the expression level of SNAI1 and SPHK2 in various cancers including human CRC and the normal adjacent tissues, RNA sequencing data from The Cancer Genome Atlas (TCGA) of colorectal adenocarcinoma was mined for SNAI1 expression values using Gene Expression Profiling Expression Analysis (GEPIA2) [39]. To find the resulting dataset, the following filters were used: gene: SNAI1; analysis type: cancer versus normal; and cancer type: colorectal adenocarcinoma. The datasets were then normalized by the trimmed mean of M values (TMM) method and recalculated for a library size of 1 million. The derived transcripts per million (TPM) values were used to measure the mRNA level of a gene for further expression stability analysis. Gene expression values were transformed as X′ = log2(X + 1), where X represents the normalized fragments per kilobase transcript per million mapped reads values. The datasets selected for analysis and visualization were the largest available datasets with complete data. The “Pathological Stage Plot” module of GEPIA2 (Gene Expression Profiling Interactive Analysis, Version 2 [40]) was used to obtain SNAI1 and SPHK2 expression in different pathological stages of colorectal adenocarcinoma within TCGA datasets.
Cell lines and culture conditions
Human CRC cell lines (HCT116, DLD1, HT29, SW480, and SW620) were obtained from ATCC (Manassas, VA) and cultured according to ATCC recommendations. The cell lines were routinely tested for mycoplasma using a polymerase chain reaction (PCR)-based detection method. HCT116 and DLD1 were transfected with empty vector pCMV-3Tag-1, pCMV-SNAI1, or empty vector pIRES-GFP and pIRES-SPHK2-GFP construct using Lipofectamine 3000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol to create overexpressing SNAI1 stable cell lines, i.e., HCT116-SN and DLD1-SN. Transfected cells were selected in a medium containing G418 (Invitrogen, Carlsbad, CA) at a final concentration of 400 µg/mL. The expressions of SNAI1 and SPHK2 were confirmed by western blot analysis.
Xenograft tumor processing and patient-derived tumor organoid (PDTO) culture
PDX creation and PDTO culture was performed as described previously [41]. Based on an Institutional Review Board (IRB) approved protocol (IRB protocol number 30678) at the Medical University of South Carolina, CRC endoscopic biopsy samples were obtained from consenting stages 2 and 3 patients before receiving neoadjuvant therapy. Briefly, a portion of the tumor tissue harvested from patients was minced with a syringe handle and then cut into < 1 mm3 tumor fragments. After washing, samples were resuspended in ammonium-chloride-potassium (ACK) lysis buffer (Fisher Scientific, Waltham, MA) before being digested with Liberase DH (Sigma-Aldrich, St. Louis, MO) containing 10 µm Y-27632 (StemRD, Burlingame, CA). The cell suspension was filtered through a 250 µm sieve followed by a 100 µm cell strainer (Fisher Scientific, Waltham, MA). Then, 5 × 105 tumor cells in 100 µL of Matrigel (Corning™) were implanted subcutaneously in the flanks of 6- to 8-week-old female NOD-SCID gamma (NSG, Jackson Laboratory, Bar Harbor, ME) mice. Tumor tissues were processed similarly to initial tumor samples. The cell suspension was collected, spun down, and resuspended in basal culture medium comprised of advanced DMEM/F12 supplemented with 10 mM HEPES, 1 × GlutaMAX, and 1 × penicillin/streptomycin (Invitrogen, Carlsbad, CA). After cell counting, tumor cells were collected for PTDO culture.
Complete organoid growth medium was made fresh using basal culture medium supplemented with 500 nM A83-01 (Sigma-Aldrich, St. Louis, MO), 1 × B27 supplement (Invitrogen, Carlsbad, CA), 50 ng/mL epidermal growth factor (Invitrogen, Carlsbad, CA), 10 nM gastrin, 1 mM N-acetyl-L-cysteine, 10 mM nicotinamide, 10 nM prostaglandin E2, 6 µM SB20219 (Sigma-Aldrich, St. Louis, MO), and 10 µM Y27632 (StemRD, Burlingame, CA). Then, 1 × 106 tumor cells were mixed with 2 mL of reduced growth factor basement membrane extract (BME) (R&D Systems, Minneapolis, MN). Cell suspensions in a complete organoid culture medium were seeded as 40 µL droplets into a prewarmed six-well plate with seven droplets per well.
Cell viability assay
Cell viability was evaluated by Cell Titer-Glo Assay (Promega, Madison, WI) according to the manufacturer’s protocol. For all monolayer cancer cells used in this study, 5,000 cells per well were plated in 96-well plates. After 24-h incubation, cells were treated with DMSO, 5FU (Sigma-Aldrich, St. Louis, MO), ABC294640, and/or S1P (Caymanchem, Ann Arbor, MI) in eight to 10 wells for 96 h.
For PDTOs, 2.5 mL of Tryple EXPRESS (Invitrogen, Carlsbad, CA) containing 10 µM Y27632 (StemRD, Burlingame, CA) was added into each well in a six-well plate. BME-containing droplets were transferred to a 15 mL conical tube and incubated at 37 °C to release the organoids from BME. In a fresh 96-well plate, 40 µL of dilute BME was placed and used as a base layer. Organoids were plated at a density of 3,000 - 4,000 cells/well in 100 µL of 2% BME in a complete organoid culture medium on the BME. PDTOs were treated in triplicates with 1 µM 5FU and 20 µM ABC294640 for 96 h. After 96-h treatment, 100 µL Cell Titer-Glo reagent was added to each well for 10 min of incubation at 37 °C. The luminescence of each well was collected, and cell viability was analyzed.
Limiting dilution spheroid assay
The ability to form spheres in a 96-well ultra-low-attachment plate was evaluated as described previously [10]. The culture medium consisted of DMEM/F12 supplemented with 1 × B27, 20 ng/mL epidermal growth factor, 20 ng/mL fibroblast growth factor basic, 0.5 mg/mL BSA, 4 mg/mL heparin, 55 mM 2-mercaptoethanol, and 1 × penicillin/streptomycin (Invitrogen, Carlsbad, CA). After 10 days of incubation, the total number of spheres greater than 50 µM in diameter was quantified under light microscopy.
siRNA transfection
SPHK2 and SNAI1 expression were downregulated by transfection with sequence-specific siRNAs. siRNA against human SPHK2 (targeted sequence: CCCUGAAACUAAACAAGCUUGGUAC, 5′ to 3′), siRNA against human SNAI1 (targeted sequence: CAACUGCAAAUACUGCAACAAGGAA, 5′ to 3′), and scrambled control siRNA (UACAGUUUAUUGAUAUUCAAUAAAG, 5′ to 3′) (IDT technologies, Coralville, IA, USA) were used. The siRNAs were diluted in 500 µL of OptiMem (Thermo Fisher Scientific, Waltham, MA, USA). Lipofectamine™ 2000 (Thermo Fisher Scientific, Waltham, MA, USA) was mixed gently before use, and the appropriate amount was subsequently diluted in 500 µL of OptiMem. The solution was incubated for 5 min at room temperature. Following 5 min of incubation, the diluted DNA was combined with diluted Lipofectamine™ 2000 (total volume 1 mL). The solution was mixed gently and incubated for 20 min at room temperature. The complexes (1 mL) were added to a 60 mm dish containing cells and medium. The cells were incubated at 37 °C in a CO2 incubator for 18 to 48 h before further assays.
Luciferase promoter assay
The luciferase reporter plasmid containing the putative 1.5-kb SPHK2 promoter in the pEZX-PG04 basic vector (GeneCopoeia, Rockville, MD, USA) was purchased from GeneCopoeia. The luciferase activity was measured using the Secrete-Pair Dual Luminescence Assay Kit (GeneCopoeia, LF031, Rockville, MD, USA) in a Clariostar microplate reader (BMG Labtech, NC, USA). Briefly, HCT-V and HCT-SN cells cultured in 24-well plates were transfected with plasmids containing pCMV-SNAI1 and/or with the pEZX-PG04-SPHK2 promoter Gaussia luciferase/secreted alkaline phosphatase plasmid (GeneCopoeia,) using Lipofectamine 2000. The medium was collected after 48 h, and the Gaussia luciferase activity was determined in the supernatant of HCT-V and HCT-SN cells by using the activity of secreted alkaline phosphatase as an internal control according to the manufacturer’s recommendations.
Nucleus/cytoplasm fractionation
All preparations were performed on ice. A sample of 1 × 106 cells was washed twice and resuspended in 1 mL ice-cold HBSS (Thermo Fisher Scientific, Waltham, MA, USA). The suspension was centrifuged at 3,000 rpm for 5 min at 4 °C, resuspended in 900 µL HBSS containing 0.1% NP-40 (Sigma-Aldrich, St. Louis, MO), and pipetted up and down five times. The suspension (300 µL) was separated, and 100 µL of 4 × Laemmli buffer (BioRad Laboratories, Hercules, CA) was added (whole-cell fraction). The remaining suspension (600 µL) was centrifuged at 3,000 rpm for 5 min at 4 °C, 300 µL of this suspension was separated, re-centrifuged at 13,000 rpm for 5 min at 4 °C, 100 µL of 4 × Laemmli buffer was added and boiled at 95 °C for 1 min (cytoplasmic fraction). The remaining suspension (300 µL) was resuspended in 1 mL ice-cold HBSS containing 0.1% NP40, centrifuged at 3,000 rpm for 5 min at 4 °C. The supernatant was discarded, and the pellet was resuspended in 180 µL of 1 × Laemmli buffer (nuclear fraction). The whole cell and nuclear fractions were sonicated for 5 s three times, and then 10 µL, 10 µL, and 5 µL of whole-cell, cytoplasmic, and nuclear fractions, respectively, were used for western blotting analysis.
Immunofluorescence staining
CRC cells grown on coverslips were fixed in 100% methanol for 10 min at room temperature and permeabilized with 0.1% Triton X-100 followed by incubation with blocking buffer (Cell Signaling Technology, Danvers, MA) for 1 h at room temperature. Next, the cells were incubated with mouse anti-E-cadherin monoclonal antibody (1:100, Santa Cruz Biotechnology, Dallas, TX) diluted in immunofluorescence antibody dilution buffer (Cell Signaling Technology, Danvers, MA) overnight at 4 °C. The cells were then washed three times in 1 × phosphate-buffered saline (PBS) for 5 min each and were incubated with Alexa 488-conjugated anti-mouse secondary antibody (Cell Signaling Technology, Danvers, MA) for 1 h at room temperature. The cells were then washed three times in 1 × PBS for 5 min each and mounted on glass slides using a mounting medium containing DAPI (Abcam, Waltham, MA). Cells were then observed under a fluorescence microscope (EVOS M5000, Thermo Fisher Scientific, Waltham, MA, USA), and pictures were taken at 40 × magnification.
Western blot analysis
Cells were suspended in RIPA protein lysis buffer (Thermo Fisher Scientific, Waltham, MA) with a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). Protein concentration was quantified using bicinchoninic acid (BCA) protein assay (Thermo Fisher Scientific, Waltham, MA). Approximately 40 - 65 µg total protein was resolved with sodium dodecyl sulfate polyacrylamide gel electrophoresis SDS-PAGE (10% polyacrylamide gel) and transferred to a nitrocellulose membrane (GE Healthcare, Marlborough, MA). Blots were probed with primary antibodies SNAI1, actin (Cell Signaling Technology, Danvers, MA), sphingosine-1-phosphate receptor (S1PR)1, S1PR2, S1PR5, SPHK2, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), lamin B1, cMyc (Proteintech, Rosemont, IL), S1PR3 (Invitrogen, Carlsbad, CA), S1PR4 (Lifespan Biosciences, Lynnwood, WA), SPHK1, beta-actin (Santa Cruz Biotechnology, Dallas, TX) and HRP-conjugated secondary antibodies (Cell Signaling Technology, Danvers, MA). Immunoblots were visualized with enhanced chemiluminescence (GE Healthcare, Marlborough, MA) by the BioRad Imaging System (BioRad Laboratories, Hercules, CA).
Lipidomics analysis
Sphingolipids in SNAI1 overexpressing stable cells (1 × 106) were measured using liquid chromatography-mass spectrometry (LCMS) by the Lipidomics Shared Resource Facility at the Medical University of South Carolina, as described previously [42]. The lipid abundance was normalized to inorganic phosphate (Pi).
Isolation of CSCs
Identification of aldehyde dehydrogenase positive (ALDH+) cells was performed using the ALDEFLUOR kit (STEMCELL Technologies, Cambridge, MA) according to the manufacturer’s instructions. Dissociated cells from the PDXs were resuspended in ALDEFLUOR assay buffer (4 × 105 cells/mL) and incubated with 1.5 mM ALDH substrate BODIPY-amino acetaldehyde. One aliquot of this cell mixture was immediately transferred to the tube containing the ALDH inhibitor (N, N-diethylamino)benzaldehyde (DEAB) to serve as the negative control. These samples were incubated for 45 min at 37 °C to allow the generation of fluorescent product. Cells were then incubated with anti-human EpCAM-PE (epithelial marker, Miltenyi Biotec, Auburn, CA) for 30 min on ice to label EpCAM cells. ALDH+ and EpCAM+ cells were sorted using FACS (BD Aria II Cell Sorter) by comparing the fluorescence of test samples against that of control samples that were treated with the ALDH inhibitor DEAB.
In vivo CRC therapy model
A CRC xenograft experiment was performed with SCID/bg mice housed in individually ventilated cages with enrichment (four mice per cage) in temperature-controlled rooms with access to water and food ad libitum, to evaluate the in vivo antitumor activity of ABC294640 combined with 5FU. SW620 cells were counted and washed once in ice-cold PBS. SW620 cells (1 × 106) in 50 µL PBS and 50 µL Matrigel basement membrane matrix (Corning, Glendale AZ) were implanted subcutaneously in the right flank of 6 - 8-week-old SCID/bg male mice (Jackson Laboratory, Sacramento, CA) under general anesthesia (2% isoflurane), and the tumor volumes were calculated using the equation: (length (L) × width (W)2)/2. When the tumors reached 100 mm3, mice were randomly assigned to four groups and received intraperitoneal (IP) injections with either vehicle control n = 6 (DMSO) (Sigma-Aldrich, St. Louis, MO), 5FU n = 6, (50 mg/kg of body weight dissolved in DMSO), ABC294640 n = 6, (75 mg/kg of body weight dissolved in DMSO), or the combination of 5FU and ABC294640 n = 6. All treatments were performed 3 days/week. Mice body weight and tumor volume measurements were performed twice every week. At the end of the experiment (at < 4 weeks after cell implantation or at 15-mm tumor diameter), mice were euthanized, and the tumors were excised for subsequent analyses.
Statistical analysis
Statistical analyses were performed using the Student’s t-test for paired data for in vitro experiments. For the in vivo experiment, statistical analyses were performed using one-way analysis of variance (ANOVA) analysis for comparison of multiple groups. P < 0.05 was considered significant.
Institutional Review Board approval
The study protocol was approved by the Institutional Review Board (IRB) at Baylor College of Medicine. The animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) of Baylor College of Medicine following national guidelines.
Ethical compliance with human/animal study
This study was conducted in compliance with the ethical standards of the responsible institution on human subjects as well as with the Helsinki Declaration.
| Results | ▴Top |
The association between SNAI1 and sphingosine kinase 2 expression in CRC
Data mined from TCGA demonstrated that RNA expression levels of SNAI1 and SPHK2 transcripts were significantly higher in tumor samples compared with normal adjacent tissues, including CRCs, esophageal, and head and neck squamous cell carcinoma (Fig. 1a). In addition, the expression of SNAI1 and SPHK2 appeared conserved across all stages of the disease (Supplementary Material 1, www.wjon.org), suggesting that these molecular mediators may play a role in cancer progression at all stages. To further explore the relationship between SNAI1 and SPHK2, we determined the expression of SPHK1, SPHK2, S1PR1-5, and SNAI1 in five established CRC cell lines as well as in SNAI1 overexpressing stable cell lines (DLD-SN, HCT-SN), compared with control empty-vector cell lines (HCT-V and DLD-V). The highly metastatic SW620 line with elevated endogenous SNAI1 showed activated sphingolipid signaling networks with upregulated SPHK2 and S1PR4 expression compared to non-metastatic CRC lines (Fig. 1b, and Supplementary Material 2, www.wjon.org). Ectopic expressing SNAI1 CRC cell lines (DLD-SN and HCT-SN) also demonstrated increased SPHK2 compared with control cells (Fig. 1b, and Supplementary Material 2, www.wjon.org). Based on densitometry, HCT-SN cells, and DLD-SN cells demonstrated a 3.2-fold and 1.7-fold increase in SPHK2 protein expression, respectively compared with control cells. SPHK2 was identified in both the cytoplasmic and nuclear cellular fractionations (Fig. 1c). In contrast, no consistent change was detected in SPHK1 levels in DLD-SN and HCT-SN cells compared with control cells (Fig. 1b, and Supplementary Material 2, www.wjon.org).
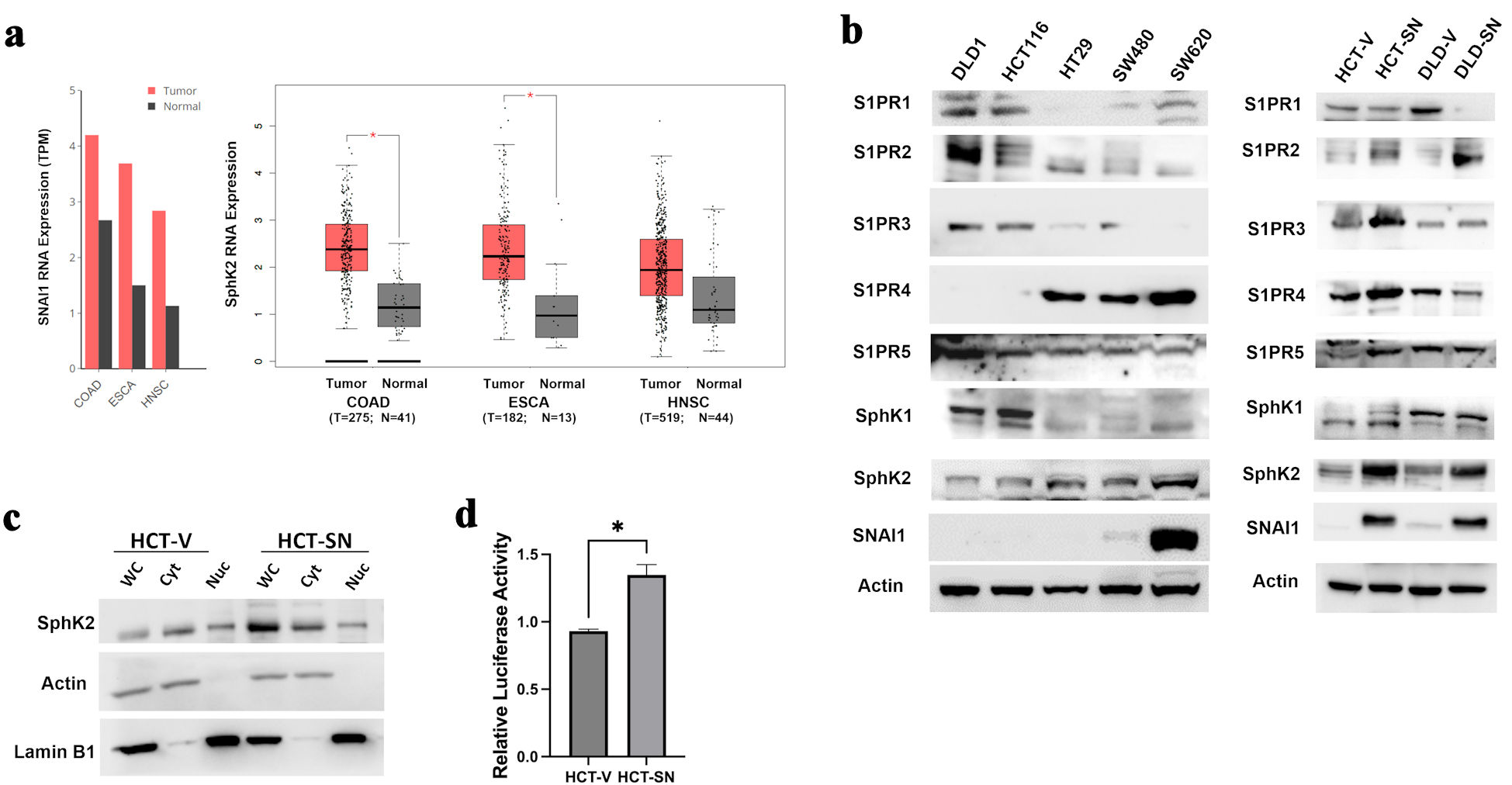 Click for large image | Figure 1. SNAI1 expression is associated with increased SPHK2 levels in CRC. (a) Comparisons of SNAI1 (left) and SPHK2 (right) RNA levels between tumoral and normal adjacent tissues in colorectal adenocarcinoma (COAD), esophageal adenocarcinoma (ESCA), and head and neck squamous cell cancer (HNSC) from TCGA datasets. Bars represent transcripts per million (TPM). Calculated means ± SEM are represented by bars and whiskers. *P < 0.05. (b) Western blot analysis for key S1P mediators in parental CRC cell lines (left panel) and ectopic-expressing SNAI1 cell lines compared with empty control vector cells (right panel). (c) Western blot analysis for SPHK2 in the cytoplasmic and nuclear fractions of HCT-SN cell line, compared with empty control vector cells. (d) SPHK2 luciferase reporter activity of HCT-SN empty vector control cells transfected with SNAI1 siRNA, compared to control siRNA treated cells. CRC: colorectal cancer; SPHK2: sphingosine kinase 2; TCGA: The Cancer Genome Atlas; SEM: standard error of the mean; S1P: sphingosine-1-phosphate; S1PR: sphingosine-1-phosphate receptor. |
S1P regulates cellular processes such as cell proliferation, differentiation, and apoptosis by binding to five different G protein-coupled receptors named S1PR1-5 [43, 44]. Interestingly, both ectopic SNAI1 cell lines demonstrated elevated S1PR2 levels. Prior reports have suggested that S1PR2 activation is associated with reduced invasion/migration, suggesting that the relationship with EMT factors requires further investigation [45]. HCT-SN and DLD-SN cells also demonstrated variable levels of S1PR1, 3, 4, 5 compared with control cells (Fig 1b, and Supplementary Material 2, www.wjon.org). Based on LCMS, ceramide levels in ectopic SNAI1 cells were comparable to control cell lines (Supplementary Material 2, www.wjon.org). Ceramide 24, one of the most abundant ceramides, was highly expressed in all cell lines tested [46].
SNAI1 induces SPHK2 promoter activity in SNAI1 overexpressing CRC cells
To determine if SNAI1 directly influences SPHK2 expression, we transfected the ectopic SNAI1-overexpressing CRC cell line HCT-SN with the luciferase reporter plasmid containing the putative SPHK2 promoter sequence. In the HCT-SN cell line, the SPHK2 promoter activity was enhanced 1.4-fold when compared with vector-transfected control (Fig. 1d). To further confirm the direct effect of SNAI1 on the promoter activity and expression of SPHK2 in CRC cells, we knocked down SNAI1 in the metastatic SW620 cell line and transfected the luciferase reporter plasmid with the putative SPHK2 promoter sequence. Compared with control siRNA-treated SW620 cells, SPHK2 promoter activity was significantly decreased by 13% in the SW620 cells transfected with SNAI1 siRNA (Supplementary Material 2, www.wjon.org).
SNAI1-driven stemness is mediated by SPHK2/S1P signaling
As SNAI1 is a well-established EMT marker, we characterized the morphologic cellular changes associated with SNAI1 expression by evaluating E-cadherin membrane expression with immunofluorescence staining. Compared with control HCT-V cells, HCT-SN cells demonstrated reduced E-cadherin expression as well as loss of membrane-bound E-cadherin (Supplementary Material 3, www.wjon.org). Similarly, DLD-SN cells also revealed a reduction in membrane-bound E-cadherin expression associated with an increased level of cytoplasmic E-cadherin compared to DLD-V cells (Supplementary Material 3, www.wjon.org). Both ectopic SNAI1 expression cell lines demonstrated loss of cell-cell contact and increased spindle shape. The morphologic changes observed in the ectopic SNAI1 expression cell lines were consistent with EMT changes when compared to the respective control cell lines.
Consistent with previous reports, ectopic SNAI1 expression resulted in enhanced cancer cell self-renewal in CRC cell lines, based on a limited dilution in vitro spheroid assay compared with control cell lines (Fig. 2a, b). In HCT-SN and DLD-SN cell lines, siRNA knockdown of SPHK2 significantly reduced spheroid generation compared with control cell lines (Fig. 2a, b, and Supplementary Material 3, www.wjon.org). Similarly, SPHK2 siRNA significantly decreased spheroid formation in parental SW620 cells (Fig. 2c, and Supplementary Material 3, www.wjon.org). SPHK2 can function through both S1P-dependent and S1P-independent downstream pathways [19]. To evaluate S1P-dependent signaling downstream of SPHK2, we assessed the ability of exogenous S1P to rescue SNAI1-mediated self-renewal following SPHK2 inhibition. Compared with vehicle and control siRNA-treated cells, exogenous S1P significantly increased spheroid formation in HCT-SN, DLD-SN, and SW620 cells transfected with SPHK2 siRNA, indicating that SPHK2 self-renewal capacity is mediated by S1P (Fig. 2a-c). Taken together, these data indicate that CRC cells with high SNAI1 acquire a CSC phenotype mediated by SPHK2-S1P signaling.
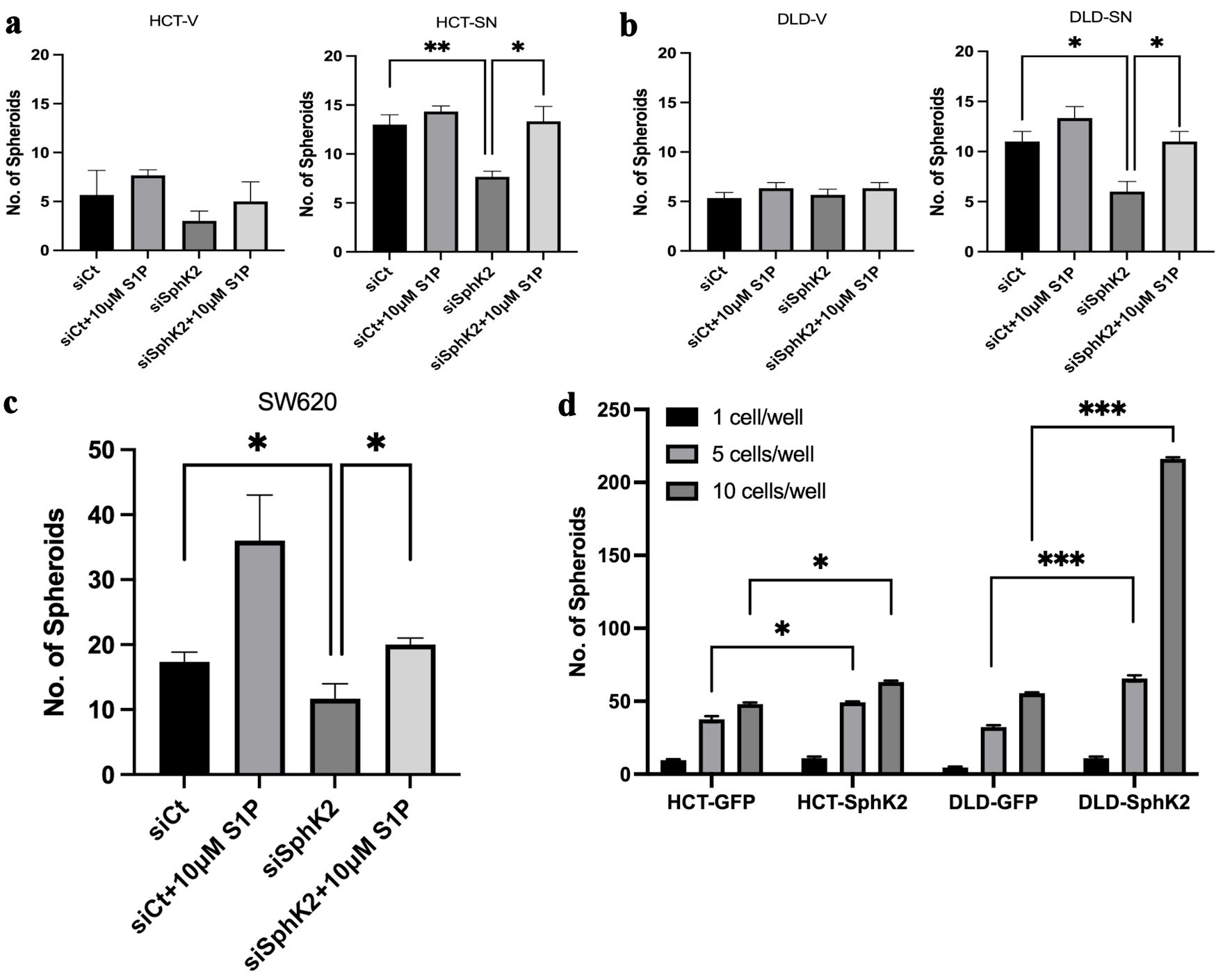 Click for large image | Figure 2. SPHK2 mediates stemness in ectopic SNAI1 expressing CRC cells. Limited dilution spheroid assay of (a) HCT-SN cells and (b) DLD-SN compared with empty vector control cells transfected with siSPHK2 or control for 10 days. (c) Limited dilution spheroid assay of SW620 cells transfected with siSPHK2 or control for 10 days. (d) Limited dilution spheroid assay comparing HCT-SPHK2 and DLD-SPHK2 cells with GFP vector (GFP) control cells. Limited dilution was performed with one cell or five cells or 10 cells per well. Data are shown as mean ± SD (*P < 0.05, **P < 0.01, ***P < 0.005). CRC: colorectal cancer; SPHK2: sphingosine kinase 2; S1P: sphingosine-1-phosphate; SD: standard deviation. |
To confirm the role of SPHK2 in CRC stemness, we ectopically expressed SPHK2 in both HCT116 and DLD-1 cells (HCT-SPHK2 and DLD-SPHK2) (Supplementary Material 3, www.wjon.org). Similar to ectopic SNAI1 expression, ectopic SPHK2 colon cancer cell lines (HCT-SPHK2 and DLD-SPHK2) demonstrated increased spheroid formation compared with control cell lines (HCT-GFP and DLD-GFP) (Fig. 2d, and Supplementary Material 3, www.wjon.org). For instance, DLD-SPHK2 cells demonstrated a four-fold increase (P < 0.001) in spheroid formation compared with control DLD-GFP cells.
SNAI1-mediated stemness can be ablated by pharmacological inhibition of SPHK2 signaling
We next evaluated whether ABC294640, the small molecular inhibitor of SPHK2, can reduce SNAI-mediated self-renewal based on in vitro spheroid formation. In both SNAI1 overexpressing cell lines, ABC294640 was able to reduce in vitro spheroid formation compared with vehicle control (Fig. 3a, b). For example, HCT-SN and DLD-SN cells demonstrated a 31% and 40% decrease (P < 0.01) in spheroid formation, respectively, compared with HCT-V and DLD-V cells (Fig. 3a, b). The addition of exogenous S1P rescued SNAI1-mediated spheroid formation in the presence of ABC294640 (Fig. 3a, b). In the SW620 cells, only partial rescue was achieved with S1P, suggesting other non-S1P dependent mechanisms downstream of SPHK2 may be involved (Fig. 3c). These data indicated that SNAI1-mediated self-renewal can be reduced by ABC294640 which was, at least in part, mediated by S1P inhibition.
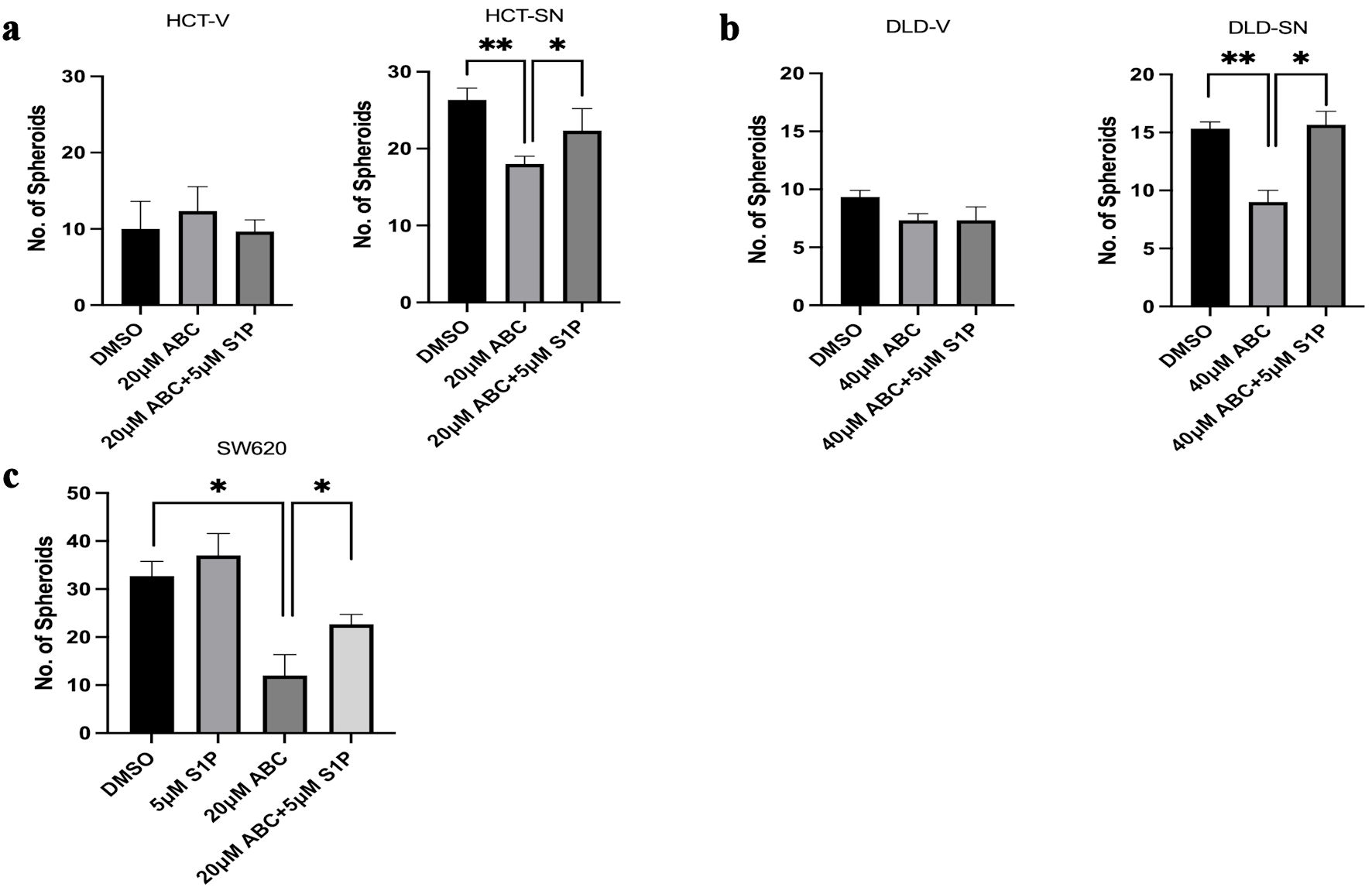 Click for large image | Figure 3. SNAI1-mediated stemness can be ablated by ABC294640. Limited dilution spheroid assay of (a) HCT-SN cells, and (b) DLD-SN cells, compared with empty vector control cells after ABC294640 treatment compared with vehicle control (DMSO). (c) Limited dilution spheroid assay of SW620 cells after ABC294640 treatment compared with vehicle control (DMSO). Rescue experiments were performed with S1P. Data are shown as mean ± SD; n = 12 (*P < 0.05, **P < 0.01). S1P: sphingosine-1-phosphate; SD: standard deviation. |
Human colorectal CSCs show elevated expression of SPHK2
ALDH activity can identify CSCs in fresh human CRC samples [47]. Our previous investigation demonstrated that EpCAM+/ALDH+ cells isolated from CRC PDXs possess in vitro and in vivo stem cell properties and high SNAI1 expression [13]. To investigate the role of SPHK2 in human CRC CSCs, we compared the expression of SPHK2 in EpCAM+/ALDH+ and EpCAM+/ALDH- cells by western blot analysis of CRC PDXs (MRC02 and MRC25), as we have previously reported [13]. EpCAM+/ALDH+ cells from both MRC02 and MRC25 PDX models expressed significantly higher SPHK2 protein compared with EpCAM+/ALDH- cancer cells. For example, MRC02 and MRC25 EpCAM+/ALDH+ cells demonstrated a nine-fold and three-fold increase in SPHK2 protein expression, respectively, compared with EpCAM+/ALDH- cancer cells (Fig. 4a-c, Supplementary Material 4, www.wjon.org). Thus, human CRC CSCs isolated by ALDH activity from PDXs demonstrate elevated expression of SPHK2 compared with expression in CRC non-CSC cells.
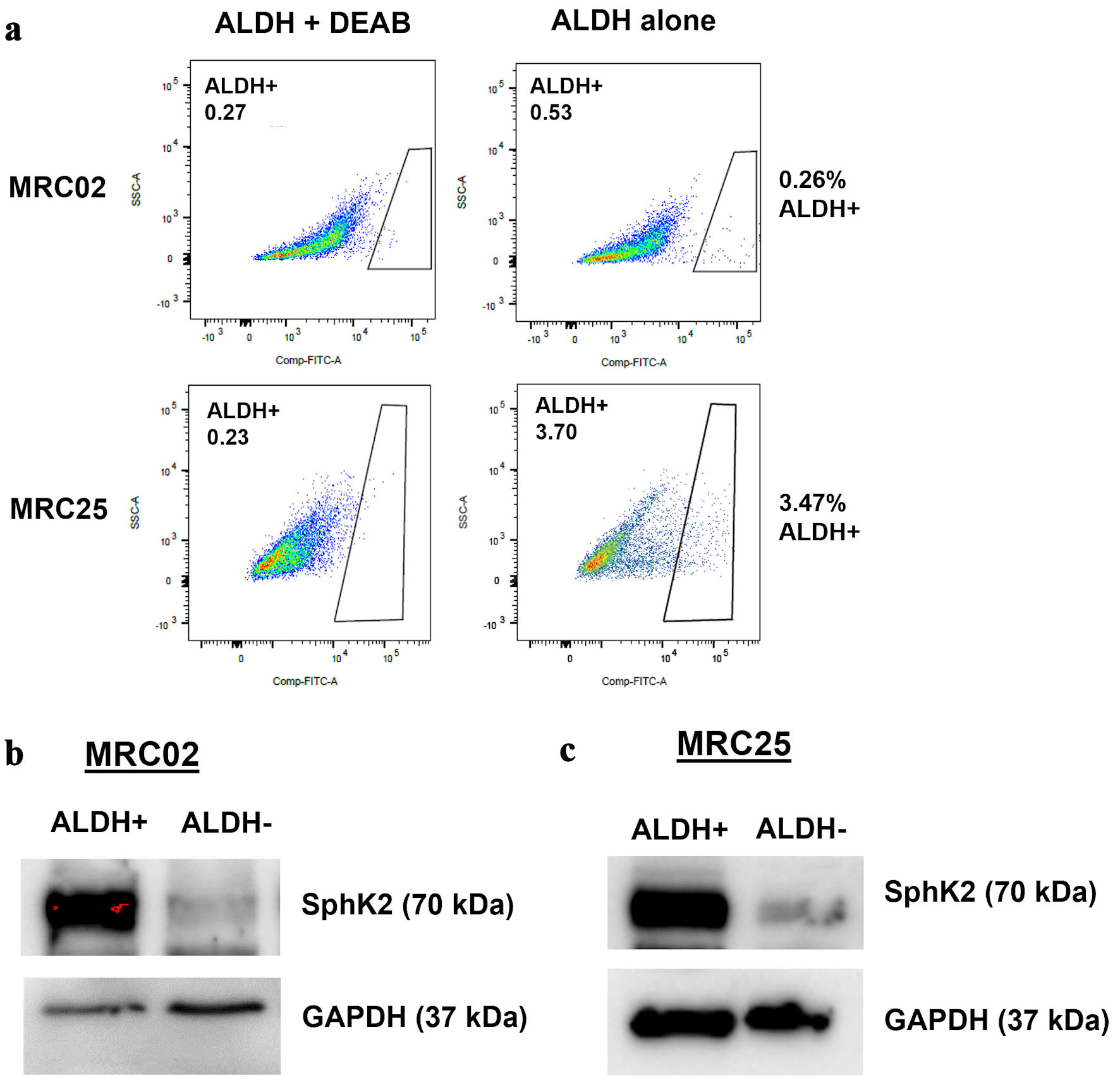 Click for large image | Figure 4. SPHK2 is overexpressed in colorectal cancer stem cells. (a) Representative flow cytometry analysis of aldehyde dehydrogenase (ALDH) activity with and without (N, N-diethylamino)benzaldehyde (DEAB), an inhibitor of ALDH in two patient-derived xenograft tumors: MRC02 and MRC25. (b, c) Western blot analysis for expression of SPHK2 in MRC02 and MRC25. SPHK2: sphingosine kinase 2; GAPDH: glyceraldehyde 3-phosphate dehydrogenase. |
SNAI1-driven 5FU chemoresistance is mediated by sphingosine kinase 2/sphingosine-1 phosphate signaling
Multiple prior studies have demonstrated that CSCs and EMT signaling mediate chemotherapy resistance [5-8], although strategies targeting these pathways have not been incorporated into clinical practice for CRC patients. Therefore, we explored SPHK2-S1P inhibition as a strategy to overcome SNAI1-mediated 5FU chemotherapy resistance. As shown in Figure 5a, b, both ectopic SNAI1 expressing CRC cell lines (HCT-SN and DLD-SN) showed reduced in vitro sensitivity to 5FU compared with control cell lines. Knockdown of SPHK2 with siRNA increased in vitro sensitivity of both HCT-SN and DLD-SN to 5FU by 1.2- and 1.9-fold (P < 0.001 and P < 0.001) at 5 µM, and 1.1- and 1.4-fold (P < 0.01 and P < 0.001) at 10 µM, respectively (Fig. 5a, b, and Supplementary Material 5, www.wjon.org). Similarly, we also observed the ability of SPHK2 inhibition to enhance 5FU sensitivity in parental SW620 (Supplementary Material 5, www.wjon.org). Conversely, exogenous S1P rescued the 5FU chemotherapy resistance in HCT-SN, DLD-SN, and SW620 cells transfected with SPHK2 siRNA (Supplementary Material 5, www.wjon.org). These results implicate S1P signaling in mediating SNAI1 chemoresistance and suggest that therapeutic targeting of S1P may enhance 5FU efficacy in CRC, especially in EMT-transformed cells.
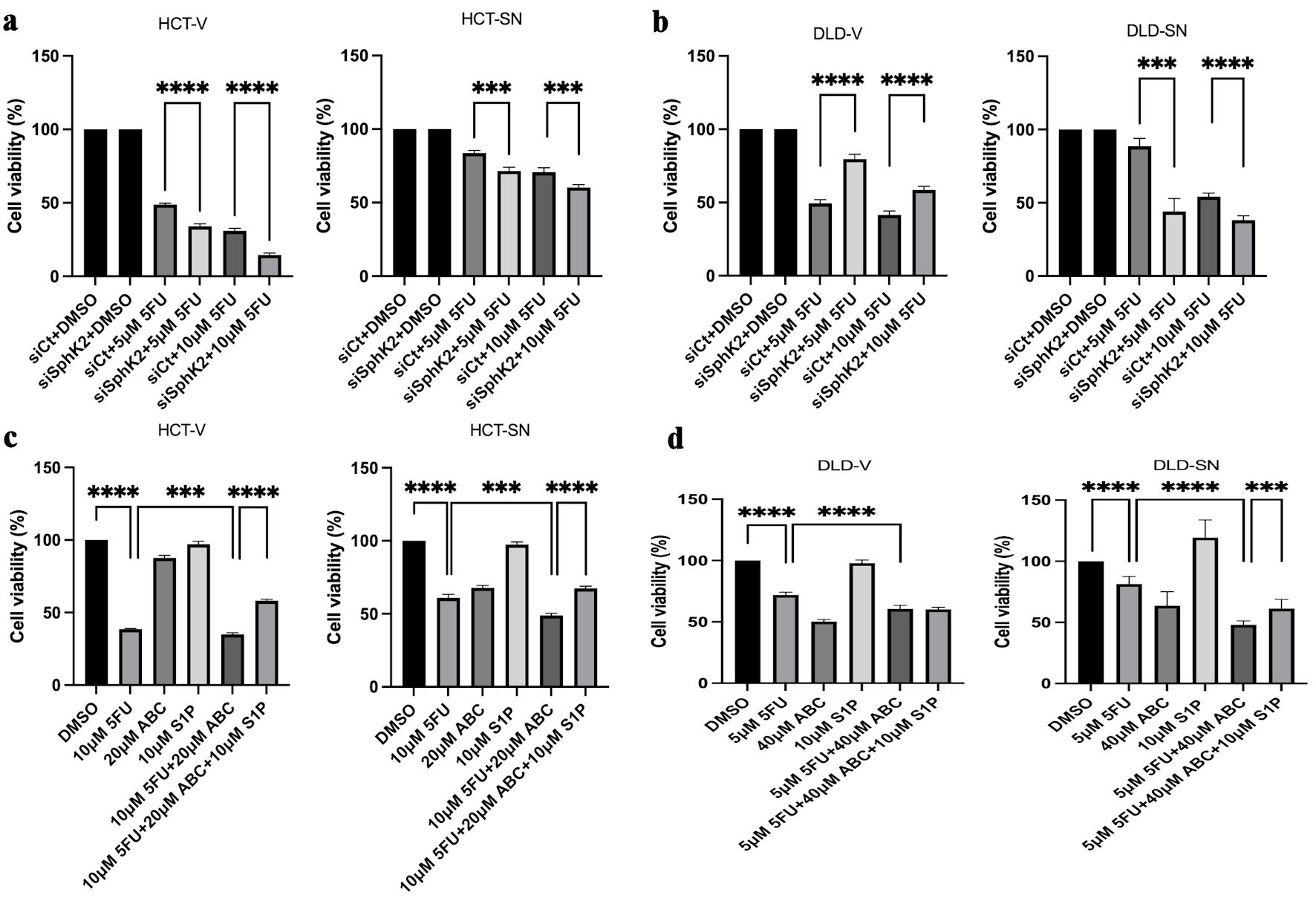 Click for large image | Figure 5. SPHK2 inhibition increases 5FU sensitivity in ectopic SNAI1 expressing CRC cells. Cell viability was assessed 96 h after 5FU treatment compared with vehicle control (DMSO) in (a) HCT-SN cells and (b) DLD-SN cells compared with empty vector control cells transfected with SPHK2 siRNA or control siRNA. Similarly, cell viability was assessed 96 h after treatment with 5FU, ABC294640, or the combination in (c) HCT-SN cells, or (d) DLD-SN cells and compared with empty vector control cells (left and right panel). Rescue experiments were performed with S1P. Data are shown as mean ± SD; n = 5 (**P < 0.01, ***P < 0.005, ****P < 0.001). CRC: colorectal cancer; SPHK2: sphingosine kinase 2; 5FU: 5-fluorouracil; SD: standard deviation. |
ABC294640 enhances 5FU chemotherapy sensitivity in high SNAI1-expressing colon cancer models
Similar to SPHK2 siRNA knockdown, ABC294640 also sensitized cells to 5FU. In the HCT-SN and DLD-SN cell lines, an additive effect was demonstrated with the combination of 5FU and ABC294640. In HCT-SN cells, the combination reduced cell viability by 37% compared with 5FU alone (P < 0.005) (Fig. 5c). The combination of 5FU and ABC294640 had a similar additive therapy effect in the DLD-SN cells leading to a reduction in cell viability by 41% compared with 5FU alone (P < 0.001) (Fig. 5d). In the parental SW620 cell line with high baseline levels of SNAI1, the combination of 5FU and ABC294640 reduced cell viability by 33% compared with 5FU alone (P < 0.005) (Supplementary Material 6, www.wjon.org). Exogenous S1P decreased the cytotoxicity of ABC294640 combined with 5FU in all cell lines tested (Fig. 5c, d, and Supplementary Material 6, www.wjon.org), suggesting that the additive therapeutic benefit of ABC294640 is at least in part mediated by inhibition of S1P.
Next, we investigated the synergistic in vivo antitumor effect of the combination of SPHK2 inhibition and 5FU in a subcutaneous xenograft model with SW620 cells (Fig. 6a). Based on tumor volume, the combination of ABC294640 and 5FU demonstrated a 69% decrease in tumor size compared with control tumors after day 18 of treatment (P < 0.05) (Fig. 6b). After 10 days of treatment and tumor harvesting, we observed a statistically significant decrease in tumor weight in the treated tumors compared with the vehicle control group (Fig. 6c). ABC294640 alone demonstrated a 48% decrease, and 5FU alone demonstrated a 42% decrease in tumor weight compared with vehicle control tumors. The combination significantly increased therapeutic efficacy compared to each therapy alone and led to a 73% decrease in tumor weight compared with vehicle control. No significant weight change was noted in the treatment groups, suggesting that the therapy combination was well tolerated (Supplementary Material 7, www.wjon.org).
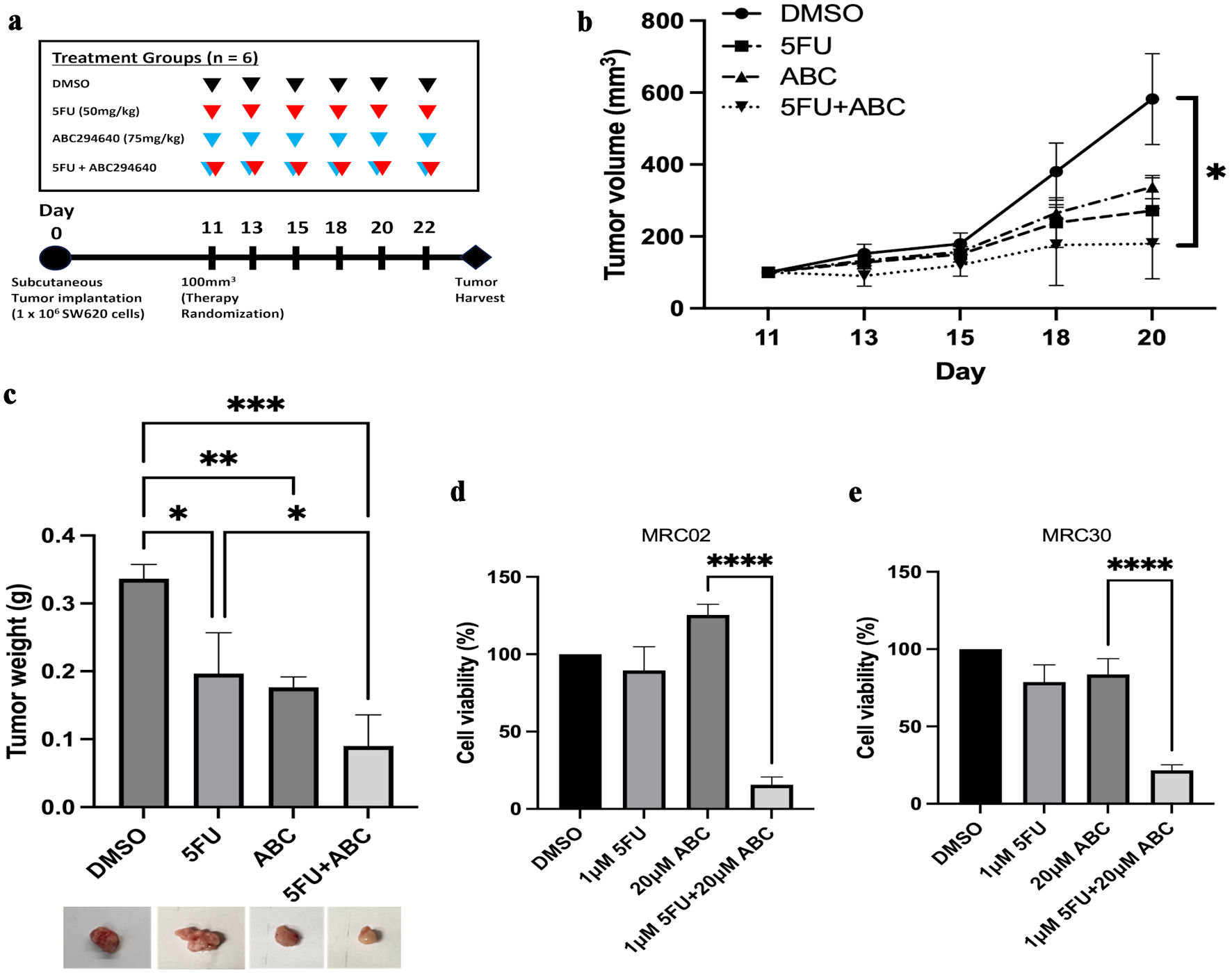 Click for large image | Figure 6. ABC294640 increases 5FU sensitivity in SW620 in vivo xenografts and CRC PDTOs. Following treatment for 11 days, subcutaneous tumors were harvested and analyzed. (a) In vivo experimental schema. After subcutaneous injection of SW620 cells, mice were randomized to four treatment groups of six mice each, starting 11 days postoperatively. 5FU (50 mg/kg) (red arrows) and 75 mg/kg of ABC294640 (blue arrows) were intraperitoneally injected three times per week. Tumors were harvested 20 - 22 days after tumor implantation. (b) Average measured tumor volume of subcutaneous SW620 xenografts throughout treatment. (c) Average tumor weight of harvested SW620 xenografts. The error bars represent mean ± SD; n = 3 (*P < 0.05, **P < 0.01, ***P < 0.005). (d, e) Cell viability was assessed 96 h after 5FU or ABC294640 treatment compared with vehicle control (DMSO) in two PDTO models: MRC02 (c), and MRC30 (d). Data are shown as mean ± SD; n = 5 (****P < 0.001). CRC: colorectal cancer; 5FU: 5-fluorouracil; SD: standard deviation; PDTOs: patient-derived tumor organoids. |
Pharmacological inhibition of SPHK2 in CRC PDTOs enhances 5FU response
To evaluate this interaction in a more translational model system, we tested the combination of ABC294640 and 5FU in therapy-resistant CRC PDTOs [41]. Two PDTO models (MRC02 and MRC30) were previously generated from CRC samples that were resistant to 5FU combined with radiation [41]. Based on cell viability assay, MRC02 and MRC30 PDTO models showed limited response to either 5FU or ABC294640 alone. In contrast, when 5FU was combined with ABC294640, both PDTO models demonstrated a significantly improved response when compared with either therapy alone or vehicle control (P < 0.001) (Fig. 6d, e). For example, the combination treatment demonstrated an 84% and 78% decrease in cell viability in MRC02 and MRC30 organoids, respectively, compared with DMSO-treated organoids. Taken together, these data indicated that SPHK2-S1P signaling can be therapeutically targeted to overcome SNAI-mediated 5FU resistance in CRC cell models and represents a promising strategy for treatment-resistant CRCs.
| Discussion | ▴Top |
Although decades of research have resulted in a better understanding of CRC pathophysiology and increased therapeutic options, chemoresistance is a common occurrence in CRC resulting in treatment failure and death. Accumulating evidence has demonstrated that a strong link exists between the development of chemoresistance, EMT, and CSCs [6, 8, 10, 12]. In particular, SNAI1 is a potent driver of stemness in CRC [48], capable of inducing EMT [10] as well as chemoresistance [49]. Further, the downstream molecular mechanisms of SNAI1 in the context of CRC are not well defined, and the ability to target EMT pathways and/or CSC remains elusive, without clinically relevant therapies. We hypothesized that SPHK2-S1P signaling may mediate SNAI1-driven therapy resistance and serve as a molecular target to improve chemotherapy response in CRC.
Growing evidence suggests that S1P supports CSCs in various cancers [50-54], including CRC [54]. The interaction between SPHK1, EMT, and metastasis in CRC has been previously studied [55-57]. For example, SPHK1 is associated with worse clinical outcomes in CRC [55, 57] and promotes CRC metastasis by inducing EMT through the FAK/AKT/MMP axis [56]. However, the role of SPHK2 is less defined. In the present study, we demonstrated the role of SNAI1/SPHK2/S1P signaling in mediating CRC chemotherapy resistance and stemness. SPHK2 expression was significantly increased and transcriptionally regulated by SNAI1 in the SNAI1-high, metastatic SW620 CRC cell line, as well as in ectopic expressing SNAI1 CRC cell lines. Notably, human CRC stem cells isolated from two colorectal patient-derived models also demonstrated significantly high expression of SPHK2. We also observed that the SNAI1-driven self-renewal in CRC is mediated by SPHK2 in an S1P-dependent fashion based on in vitro limited dilution spheroid formation. Taken together, these data suggest that SPHK2 mediates SNAI1-driven stemness through S1P signaling. Our evidence supports SPHK2/ S1P as a novel mediator of CRC stem cell phenotype, especially considering the high expression in the human CRC stem cells isolated from CRC PDXs. Importantly, the next steps would be to prospectively analyze S1P signaling within fresh tumor samples from treatment-refractory CRC patients using techniques such as liquid chromatography-mass spectrometry [58].
Chemoresistance remains a central issue in CRC [59], and efforts to resensitize cancer cells to chemotherapy agents is an active area of investigation [60, 61]. The present study demonstrated that the knockdown of SPHK2 by either siRNA or pharmacological inhibition increased 5FU sensitivity in the therapy-resistant SNAI1-high CRC cell lines. ABC294640 is an attractive drug for SPHK2 inhibition, as it is administered orally, well tolerated in patients, and demonstrated antitumor activity in a phase 1 clinical trial [29]. Encouragingly, the combination of ABC294640 and 5FU successfully overcame 5FU drug resistance in both in vivo CRC xenografts and in two CRC patient-derived organoid models from therapy-resistant patients. Of note, previous studies have reported a positive correlation between SNAI1 and programmed death-ligand 1 (PDL1) expression [62], and SNAI1 has also been shown to promote immunosuppression via the recruitment of myeloid-derived suppressor cells and production of immunosuppressive natural T-regulatory like CD4+CD25- cells in various cancers [63], indicating that PDL1-targeted immunotherapy in combination with SPHK2 blockade may be an attractive combination for therapy-resistant CRCs. Collectively, these observations support the concept that SPHK2-S1P signaling is a viable anti-neoplastic target in high SNAI1 CRCs and a viable therapeutic strategy to overcome CRC drug resistance.
Conclusions
Therapy-resistant EMT pathways and CSC remain promising yet elusive therapy targets for CRC to improve treatment response and overcome chemotherapy resistance. Improving our understanding of the molecular pathways supporting CRC CSC represents a strategy to identify novel therapy targets. Our current work implicates SPHK2-S1P signaling as a key mediator of CRC CSCs and EMT signaling through SNAI1. These new insights suggest that targeting SPHK2-S1P with ABC294640 can serve as a novel therapeutic strategy for the treatment refractory CRC.
| Supplementary Material | ▴Top |
Suppl 1. SNAI1 and SPHK2 expression in colorectal cancer.
Suppl 2. Ceramides and sphingosine-1-phosphate receptor levels in colorectal cancer cells.
Suppl 3. SPHK2 mediates stemness in colorectal cancer cells.
Suppl 4. SPHK2 expression is elevated in human colorectal cancer stem cells.
Suppl 5. SNAI1-SPHK2 axis promotes chemoresistance in colorectal cancer cells.
Suppl 6. SNAI1-SPHK2 axis promotes chemoresistance in colorectal cancer cells.
Suppl 7. Mouse body weight over the duration of SW620 in vivo experiment.
Acknowledgments
None to declare.
Financial Disclosure
This work was supported by the National Institutes of Health (CA214461, DE016572, AG069769, and P01 CA203628 to BO) and Merit Review Award # I01 CX001880-01A1 from the United States Department of Veterans Affairs Biomedical Laboratory Research and Development Program (ERC).
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
A written informed consent was obtained from study participants.
Author Contributions
ERC: study concept and design. HJ, YZ, ZG, JD, and SAB performed the research. HJ, ZG, ERC: writing of original draft, review, and editing. HJ and ERC analyzed data and interpreted results. HJ, ZG, AJ, BO, and ERC contributed to review and revisions. The final manuscript has been approved by all authors. The work reported in the paper has been performed by the authors unless specified in the text.
Data Availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
| References | ▴Top |
- Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet. 2019;394(10207):1467-1480.
doi pubmed - Siegel RL, Wagle NS, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics, 2023. CA Cancer J Clin. 2023;73(3):233-254.
doi pubmed - Showalter SL, Showalter TN, Witkiewicz A, Havens R, Kennedy EP, Hucl T, Kern SE, et al. Evaluating the drug-target relationship between thymidylate synthase expression and tumor response to 5-fluorouracil. Is it time to move forward? Cancer Biol Ther. 2008;7(7):986-994.
doi pubmed pmc - Wei L, Wang X, Lv L, Zheng Y, Zhang N, Yang M. The emerging role of noncoding RNAs in colorectal cancer chemoresistance. Cell Oncol (Dordr). 2019;42(6):757-768.
doi pubmed - Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105-111.
doi pubmed - Nunes T, Hamdan D, Leboeuf C, El Bouchtaoui M, Gapihan G, Nguyen TT, Meles S, et al. Targeting cancer stem cells to overcome chemoresistance. Int J Mol Sci. 2018;19(12):4036.
doi pubmed pmc - Zhou Y, Xia L, Wang H, Oyang L, Su M, Liu Q, Lin J, et al. Cancer stem cells in progression of colorectal cancer. Oncotarget. 2018;9(70):33403-33415.
doi pubmed pmc - Cammareri P, Scopelliti A, Todaro M, Eterno V, Francescangeli F, Moyer MP, Agrusa A, et al. Aurora-a is essential for the tumorigenic capacity and chemoresistance of colorectal cancer stem cells. Cancer Res. 2010;70(11):4655-4665.
doi pubmed - Li N, Babaei-Jadidi R, Lorenzi F, Spencer-Dene B, Clarke P, Domingo E, Tulchinsky E, et al. An FBXW7-ZEB2 axis links EMT and tumour microenvironment to promote colorectal cancer stem cells and chemoresistance. Oncogenesis. 2019;8(3):13.
doi pubmed pmc - Fan F, Samuel S, Evans KW, Lu J, Xia L, Zhou Y, Sceusi E, et al. Overexpression of snail induces epithelial-mesenchymal transition and a cancer stem cell-like phenotype in human colorectal cancer cells. Cancer Med. 2012;1(1):5-16.
doi pubmed pmc - Supernat A, Lapinska-Szumczyk S, Majewska H, Gulczynski J, Biernat W, Wydra D, Zaczek AJ. Epithelial-mesenchymal transition and cancer stem cells in endometrial cancer. Anticancer Res. 2013;33(12):5461-5469.
pubmed - Hwang WL, Yang MH, Tsai ML, Lan HY, Su SH, Chang SC, Teng HW, et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology. 2011;141(1):279-291.e5.
doi pubmed - Zhu Y, Wang C, Becker SA, Hurst K, Nogueira LM, Findlay VJ, Camp ER. miR-145 Antagonizes SNAI1-mediated stemness and radiation resistance in colorectal cancer. Mol Ther. 2018;26(3):744-754.
doi pubmed pmc - Chu E, Koeller DM, Johnston PG, Zinn S, Allegra CJ. Regulation of thymidylate synthase in human colon cancer cells treated with 5-fluorouracil and interferon-gamma. Mol Pharmacol. 1993;43(4):527-533.
pubmed - Ciszewski WM, Chmielewska-Kassassir M, Wozniak LA, Sobierajska K. Thymidylate synthase overexpression drives the invasive phenotype in colon cancer cells. Biomedicines. 2022;10(6):1267.
doi pubmed pmc - Spiegel S, Milstien S. Sphingosine 1-phosphate, a key cell signaling molecule. J Biol Chem. 2002;277(29):25851-25854.
doi pubmed - Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381(6585):800-803.
doi pubmed - Camp ER, Patterson LD, Kester M, Voelkel-Johnson C. Therapeutic implications of bioactive sphingolipids: A focus on colorectal cancer. Cancer Biol Ther. 2017;18(9):640-650.
doi pubmed pmc - Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer. 2018;18(1):33-50.
doi pubmed pmc - Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, Milstien S, et al. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J Biol Chem. 2000;275(26):19513-19520.
doi pubmed - Hatoum D, Haddadi N, Lin Y, Nassif NT, McGowan EM. Mammalian sphingosine kinase (SphK) isoenzymes and isoform expression: challenges for SphK as an oncotarget. Oncotarget. 2017;8(22):36898-36929.
doi pubmed pmc - Diaz Escarcega R, McCullough LD, Tsvetkov AS. The functional role of sphingosine kinase 2. Front Mol Biosci. 2021;8:683767.
doi pubmed pmc - Panneer Selvam S, De Palma RM, Oaks JJ, Oleinik N, Peterson YK, Stahelin RV, Skordalakes E, et al. Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci Signal. 2015;8(381):ra58.
doi pubmed pmc - Zhang YH, Shi WN, Wu SH, Miao RR, Sun SY, Luo DD, Wan SB, et al. SphK2 confers 5-fluorouracil resistance to colorectal cancer via upregulating H3K56ac-mediated DPD expression. Oncogene. 2020;39(29):5214-5227.
doi pubmed - Gestaut MM, Antoon JW, Burow ME, Beckman BS. Inhibition of sphingosine kinase-2 ablates androgen resistant prostate cancer proliferation and survival. Pharmacol Rep. 2014;66(1):174-178.
doi pubmed - Liu W, Ning J, Li C, Hu J, Meng Q, Lu H, Cai L. Overexpression of Sphk2 is associated with gefitinib resistance in non-small cell lung cancer. Tumour Biol. 2016;37(5):6331-6336.
doi pubmed - Antoon JW, White MD, Slaughter EM, Driver JL, Khalili HS, Elliott S, Smith CD, et al. Targeting NFkB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol Ther. 2011;11(7):678-689.
doi pubmed pmc - Shi W, Zhang S, Ma D, Yan D, Zhang G, Cao Y, Wang Z, et al. Targeting SphK2 reverses acquired resistance of regorafenib in hepatocellular carcinoma. Front Oncol. 2020;10:694.
doi pubmed pmc - Britten CD, Garrett-Mayer E, Chin SH, Shirai K, Ogretmen B, Bentz TA, Brisendine A, et al. A phase I study of ABC294640, a first-in-class sphingosine kinase-2 inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2017;23(16):4642-4650.
doi pubmed pmc - Dai L, Smith CD, Foroozesh M, Miele L, Qin Z. The sphingosine kinase 2 inhibitor ABC294640 displays anti-non-small cell lung cancer activities in vitro and in vivo. Int J Cancer. 2018;142(10):2153-2162.
doi pubmed pmc - Beljanski V, Lewis CS, Smith CD. Antitumor activity of sphingosine kinase 2 inhibitor ABC294640 and sorafenib in hepatocellular carcinoma xenografts. Cancer Biol Ther. 2011;11(5):524-534.
doi pubmed pmc - Schrecengost RS, Keller SN, Schiewer MJ, Knudsen KE, Smith CD. Downregulation of Critical Oncogenes by the Selective SK2 Inhibitor ABC294640 Hinders Prostate Cancer Progression. Mol Cancer Res. 2015;13(12):1591-1601.
doi pubmed pmc - Ding X, Zhang Y, Huang T, Xu G, Peng C, Chen G, Kong B, et al. Targeting sphingosine kinase 2 suppresses cell growth and synergizes with BCL2/BCL-XL inhibitors through NOXA-mediated MCL1 degradation in cholangiocarcinoma. Am J Cancer Res. 2019;9(3):546-561.
pubmed pmc - Xun C, Chen MB, Qi L, Tie-Ning Z, Peng X, Ning L, Zhi-Xiao C, et al. Targeting sphingosine kinase 2 (SphK2) by ABC294640 inhibits colorectal cancer cell growth in vitro and in vivo. J Exp Clin Cancer Res. 2015;34(1):94.
doi pubmed pmc - Chumanevich AA, Poudyal D, Cui X, Davis T, Wood PA, Smith CD, Hofseth LJ. Suppression of colitis-driven colon cancer in mice by a novel small molecule inhibitor of sphingosine kinase. Carcinogenesis. 2010;31(10):1787-1793.
doi pubmed pmc - He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509-1512.
doi pubmed - Lewis CS, Voelkel-Johnson C, Smith CD. Suppression of c-Myc and RRM2 expression in pancreatic cancer cells by the sphingosine kinase-2 inhibitor ABC294640. Oncotarget. 2016;7(37):60181-60192.
doi pubmed pmc - Venant H, Rahmaniyan M, Jones EE, Lu P, Lilly MB, Garrett-Mayer E, Drake RR, et al. The sphingosine kinase 2 inhibitor ABC294640 reduces the growth of prostate cancer cells and results in accumulation of dihydroceramides in vitro and in vivo. Mol Cancer Ther. 2015;14(12):2744-2752.
doi pubmed pmc - Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98-W102.
doi pubmed pmc - http://gepia2.cancer-pku.cn/#analysis.
- Janakiraman H, Zhu Y, Becker SA, Wang C, Cross A, Curl E, Lewin D, et al. Modeling rectal cancer to advance neoadjuvant precision therapy. Int J Cancer. 2020;147(5):1405-1418.
doi pubmed - Panneer Selvam S, Roth BM, Nganga R, Kim J, Cooley MA, Helke K, Smith CD, et al. Balance between senescence and apoptosis is regulated by telomere damage-induced association between p16 and caspase-3. J Biol Chem. 2018;293(25):9784-9800.
doi pubmed pmc - Pyne NJ, Pyne S. Sphingosine 1-phosphate receptor 1 signaling in mammalian cells. Molecules. 2017;22(3):344.
doi pubmed pmc - Kihara Y, Maceyka M, Spiegel S, Chun J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br J Pharmacol. 2014;171(15):3575-3594.
doi pubmed pmc - Yan J, Chen Y, Wu Q, Shao L, Zhou X. Expression of sphingosine-1-phosphate receptor 2 is correlated with migration and invasion of human colon cancer cells: A preliminary clinical study. Oncol Lett. 2022;24(1):241.
doi pubmed pmc - Markowski AR, Blachnio-Zabielska AU, Guzinska-Ustymowicz K, Markowska A, Pogodzinska K, Roszczyc K, Zinczuk J, et al. Ceramides profile identifies patients with more advanced stages of colorectal cancer. Biomolecules. 2020;10(4):632.
doi pubmed pmc - Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H, Fields JZ, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009;69(8):3382-3389.
doi pubmed pmc - Roy HK, Smyrk TC, Koetsier J, Victor TA, Wali RK. The transcriptional repressor SNAIL is overexpressed in human colon cancer. Dig Dis Sci. 2005;50(1):42-46.
doi pubmed - Wang H, Li JM, Wei W, Yang R, Chen D, Ma XD, Jiang GM, et al. Regulation of ATP-binding cassette subfamily B member 1 by Snail contributes to chemoresistance in colorectal cancer. Cancer Sci. 2020;111(1):84-97.
doi pubmed pmc - Wang YC, Tsai CF, Chuang HL, Chang YC, Chen HS, Lee JN, Tsai EM. Benzyl butyl phthalate promotes breast cancer stem cell expansion via SPHK1/S1P/S1PR3 signaling. Oncotarget. 2016;7(20):29563-29576.
doi pubmed pmc - Hii LW, Chung FF, Mai CW, Yee ZY, Chan HH, Raja VJ, Dephoure NE, et al. Sphingosine Kinase 1 regulates the survival of breast cancer stem cells and non-stem breast cancer cells by suppression of STAT1. Cells. 2020;9(4):886.
doi pubmed pmc - Hirata N, Yamada S, Shoda T, Kurihara M, Sekino Y, Kanda Y. Sphingosine-1-phosphate promotes expansion of cancer stem cells via S1PR3 by a ligand-independent Notch activation. Nat Commun. 2014;5:4806.
doi pubmed - Xu J, Zhou L, Du X, Qi Z, Chen S, Zhang J, Cao X, et al. Transcriptome and lipidomic analysis suggests lipid metabolism reprogramming and upregulating SPHK1 promotes stemness in pancreatic ductal adenocarcinoma stem-like cells. Metabolites. 2023;13(11):1132.
doi pubmed pmc - Zeng S, Liang Y, Hu H, Wang F, Liang L. Endothelial cell-derived S1P promotes migration and stemness by binding with GPR63 in colorectal cancer. Pathol Res Pract. 2022;240:154197.
doi pubmed - Bae GE, Do SI, Kim K, Park JH, Cho S, Kim HS. Increased sphingosine kinase 1 expression predicts distant metastasis and poor outcome in patients with colorectal cancer. Anticancer Res. 2019;39(2):663-670.
doi pubmed - Liu SQ, Xu CY, Wu WH, Fu ZH, He SW, Qin MB, Huang JA. Sphingosine kinase 1 promotes the metastasis of colorectal cancer by inducing the epithelial-mesenchymal transition mediated by the FAK/AKT/MMPs axis. Int J Oncol. 2019;54(1):41-52.
doi pubmed pmc - Long J, Xie Y, Yin J, Lu W, Fang S. SphK1 promotes tumor cell migration and invasion in colorectal cancer. Tumour Biol. 2016;37(5):6831-6836.
doi pubmed - Schmidt H, Schmidt R, Geisslinger G. LC-MS/MS-analysis of sphingosine-1-phosphate and related compounds in plasma samples. Prostaglandins Other Lipid Mediat. 2006;81(3-4):162-170.
doi pubmed - Van der Jeught K, Xu HC, Li YJ, Lu XB, Ji G. Drug resistance and new therapies in colorectal cancer. World J Gastroenterol. 2018;24(34):3834-3848.
doi pubmed pmc - Lei X, He Q, Li Z, Zou Q, Xu P, Yu H, Ding Y, et al. Cancer stem cells in colorectal cancer and the association with chemotherapy resistance. Med Oncol. 2021;38(4):43.
doi pubmed - Blondy S, David V, Verdier M, Mathonnet M, Perraud A, Christou N. 5-Fluorouracil resistance mechanisms in colorectal cancer: From classical pathways to promising processes. Cancer Sci. 2020;111(9):3142-3154.
doi pubmed pmc - Kim S, Koh J, Kim MY, Kwon D, Go H, Kim YA, Jeon YK, et al. PD-L1 expression is associated with epithelial-to-mesenchymal transition in adenocarcinoma of the lung. Hum Pathol. 2016;58:7-14.
doi pubmed - Tang X, Sui X, Weng L, Liu Y. SNAIL1: linking tumor metastasis to immune evasion. Front Immunol. 2021;12:724200.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.