

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 15, Number 4, August 2024, pages 695-710

Identification and Validation of a Novel Tertiary Lymphoid Structures-Related Prognostic Gene Signature in Hepatocellular Carcinoma
Yin Liua, b, Chao Bo Lia, b, Yun Peng Zhaia, Shao Kang Zhanga, Ding Yang Lia, Zhi Qiang Gaoa, c, Ruo Peng Lianga, c
aDepartment of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
bYin Liu and Chao Bo Li contributed equally to this work.
cCorresponding Author: Zhi Qiang Gao and Ruo Peng Liang, Department of Hepatobiliary and Pancreatic Surgery, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Chinaand
Manuscript submitted May 11, 2024, accepted June 12, 2024, published online July 5, 2024
Short title: TLS-Related Prognostic Gene Signature in HCC
doi: https://doi.org/10.14740/wjon1893
| Abstract | ▴Top |
Background: Hepatocellular carcinoma (HCC) is one of the most common malignant tumors originating from the digestive system. Tertiary lymphoid structures (TLS), non-lymphoid tissues outside of the lymphoid organs, are closely connected to chronic inflammation and tumorigenesis. However, the detailed relationship between TLS and HCC prognosis remained unclear. In this study, we aimed to construct a TLS-related gene signature for predicting the prognosis of HCC patients.
Methods: The Cancer Genome Atlas (TCGA) clinical data from 369 HCC tissues and 50 normal liver tissues were utilized to examine the differential expression of TLS-related genes. Based on least absolute shrinkage and selection operator (LASSO) Cox regression analysis, the prognostic model was constructed using the TCGA cohort and validated in the GSE14520 cohort and International Cancer Genome Consortium (ICGC) cohort. The Kaplan-Meier (KM) and receiver operating characteristic (ROC) curves were employed to validate the predictive ability of the prognostic model. Furthermore, Cox regression analysis was applied to identify whether the TLS score could be employed as an independent prognosis factor. A nomogram was developed to predict the survival probability of HCC patients. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were performed for TLS-related genes. Genetic mutation analysis, the CIBERSORT algorithm, and single-sample gene set enrichment analysis (ssGSEA) were used to assess the tumor mutation landscape and immune infiltration. Finally, the role of the TLS score in HCC therapy was investigated.
Results: Six genes were included in the construction of our prognostic model (CETP, DNASE1L3, PLAC8, SKAP1, C7, and VNN2), and we validated its accuracy. Survival analysis showed that patients in the high-TLS score group had a significantly better overall survival than those in the low-TLS score group. Univariate, multivariate Cox regression analysis and the establishment of a nomogram indicated that the TLS score could independently function as a potential prognostic marker. A significant association between TLS score and immunity was revealed by an analysis of gene alterations and immune cell infiltration. In addition, two subtypes of the TLS score could accurately predict the effectiveness of sorafenib, transcatheter arterial chemoembolization (TACE), and immunotherapy in HCC patients.
Conclusion: In this research, we conducted and validated a prognostic model associated with TLS that may be helpful for predicting clinical outcomes and treatment responsiveness for HCC patients.
Keywords: Hepatocellular carcinoma; Tertiary lymphoid structures; Prognostic model; Therapy response
| Introduction | ▴Top |
Liver cancer ranks as the sixth most prevalent malignancy worldwide, with projections estimating over 1 million new cases by 2025 [1]. Hepatocellular carcinoma (HCC) is the predominant form of liver cancer, accounting for over 90% of occurrences, and represents a significant cause of cancer-related mortality globally [2]. Several risk factors, including dietary toxins and chronic hepatitis B and C, metabolic liver disease (especially nonalcoholic fatty liver disease), and alcohol addiction, are associated with an elevated risk of developing HCC [3]. Despite significant advancements in surgical techniques, chemotherapy, and targeted therapy for HCC treatment, some patients still suffer from metastasis and recurrence after undergoing these curative procedures [4, 5]. Consequently, there is an urgent need to develop an effective prognostic model, given the significant incidence and recurrence of HCC.
Tertiary lymphoid structures (TLS) are ectopic lymphoid organs that arise in non-lymphoid tissues at areas of chronic inflammation, such as malignancies. Their growth resembles lymphoid neogenesis, which takes place in peripheral immunization tissues after long-term exposure to inflammatory signals carried by chemokines and cytokines [6]. During the process of development, T-cell zones and B-cell zones, which contain germinal centers (GCs), aggregate together to form these multicellular and organized structures [7-9]. Furthermore, mature dendritic cells (DCs) and high endothelial venules (HEVs) are also found within TLS, which are identical to those that allow white blood cells to enter secondary lymphoid organs (SLOs) [10, 11]. Increasingly, researchers are focusing on investigating TLS in various malignancies, including HCC. Previous studies have demonstrated that TLS density correlates with the density of CD8+ T cells and CD4+ T cells within tumors, which are associated with improved prognosis in most solid malignancies [8]. Moreover, TLS may serve as markers for evaluating the efficacy of immunotherapy in treating non-small cell lung cancer (NSCLC) [12, 13]. Based on these findings, transcriptome analyses of TLS in various malignant tumors were conducted. For example, a gene signature associated with TLS was found based on the differences in gene expression between different melanoma tissues. Their research revealed that the TLS gene signature in melanoma patients was related to the clinical outcomes of immune checkpoint blockade therapy [14]. However, a reliable transcriptional-level predictive model of TLS in HCC has not yet been developed. We thus speculated that the gene signature would be able to forecast the prognosis of HCC patients.
Our research aimed to determine whether the TLS-related gene signature could serve as a biomarker to assess the prognosis of HCC patients. In this study, we constructed a prognostic model and investigated the connection between the prognostic model and clinical characteristics. Our study also demonstrated that our prognostic model is highly correlated with the tumor microenvironment (TME) and can accurately predict HCC patients’ responses to relevant therapies.
| Materials and Methods | ▴Top |
Public data acquisition and processing
The RNA sequencing (RNA-seq) data for 369 HCC samples and 50 normal liver samples, along with their clinicopathological features, were retrieved from The Cancer Genome Atlas (TCGA). The microarray data and corresponding clinicopathologic information for GSE14520, GSE104580, and GSE109211 datasets were downloaded from the Gene Expression Omnibus (GEO) portal. The Liver Cancer-RIKEN, Japan (LIRI-JP) RNA-seq data and their corresponding survival information were downloaded from the International Cancer Genome Consortium (ICGC) portal. The “limma” R package was used to standardize the data on gene expression. No ethics approval is needed.
Identification of differentially expressed TLS-related genes
The differentially expressed TLS-related genes in 369 HCC cases and 50 normal samples were found using the “DESeq2” package. As cut-off values, P < 0.05 and |log2 fold change (FC)| > 1.2 were used [15].
Enrichment analysis of differentially expressed TLS-related genes
The “clusterProfiler” package was used to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment analysis using differentially expressed TLS-related genes. Subsequently, after genetic differential analysis of HCC patients in the high- and low-TLS score groups, all genes were arranged in descending order according to Log FC, and further gene set enrichment analysis (GSEA) was performed for biological processes and signaling pathways of genes in both groups [16]. The C5 subcollection served as the source for our reference gene sets (c5.all.v7.0.entrez.gmt). We used 1,000 permutation analyses to establish the significance thresholds, and outcomes that had a P-value of less than 0.05 were deemed significant.
Construction of a prognostic model based on TLS-related genes
The TLS score was determined based on the standardized LIHC mRNA expression data and its corresponding regression coefficients. TLS score= ∑inxiyi [15]. In the aforementioned formula, “x” stands for the LASSO Cox regression analysis coefficient of TLS-related genes, and “y” stands for the gene expression of TLS-related genes. Based on the median TLS score, HCC patients were classified into high-risk and low-risk groups, and the difference in overall survival (OS) between the two groups was examined. The “timeROC” package generated receiver operating characteristic (ROC) curves to assess the prognostic effectiveness of the model.
Validating the performance of the prognostic model
We calculated the TLS score for each sample in the GSE14520 and LIRI-JP cohorts using the formula obtained from the training set. After dividing HCC patients into high- and low-TLS score groups based on median values, Kaplan-Meier (KM) survival curves were used to detect prognostic values. Subsequently, univariate and multifactorial Cox regression analyses were used to test whether the model was an independent prognostic factor for OS in HCC patients. The 1-, 3-, and 5-year ROC curves and area under the curve (AUC) values were used to evaluate the performance of the model in the validation set.
Construction of nomogram and calibration curves
To better serve clinical practice, we constructed a nomogram using the “rms” package, combining the prognostic model with important clinical traits such as age, gender, stage, and grade. The performance of the nomogram was evaluated by the calibration curves of the training and validation sets of 1, 3, and 5 years.
Genetic mutation analysis
The mutation data of the TCGA-LIHC cohort (workflow type: varScan2 variant aggregation and masking) were downloaded from its portal. The waterfall function of the “maftools” package was used to show the mutation landscape of HCC patients in the high- and low-TLS score groups.
Immune profile analysis
The proportion of the 22 types of immune cells within the TME of each sample was evaluated via the CIBERSORT algorithm in R software [17]. Single-sample gene set enrichment analysis (ssGSEA) was used to calculate the proportions of 28 different types of immune cells in the TME [18].
Sorafenib, TACE and immunotherapy sensitivity analysis
The response of HCC patients to sorafenib and TACE therapy regimens was assessed in the GSE109211 and GSE104580 cohorts using ROC curves and AUC. The sensitivity to immunotherapy in the patients with low and high TLS scores was assessed using the Tumor Immune Dysfunction Exclusion (TIDE) website [19].
Statistical analysis
The data analysis and visualization were conducted in R (version 4.2.1). Categorical variables were tested using the Chi-square test. Continuous variables were tested using the t-test or Wilcoxon sum test. The Chi-square test was used to examine data from columnar tables. The analysis of variance (ANOVA) test was used to compare data across three or more groups. Correlation analysis was performed using the Spearman method. The package “maftools” in the R language was used to determine the tumor mutation burden (TMB) score. A P-value of 0.05 or lower was regarded as statistically significant.
| Results | ▴Top |
Functional enrichment analysis of TLS-related genes
The elaborate procedure of our research is presented in Figure 1. To more clearly illustrate the biological roles of genes connected to TLS, GO and KEGG pathway enrichment analyses were performed. In particular, leukocyte cell-cell adhesion, lymphocyte differentiation, the antigen receptor-mediated signaling pathway, regulation of T-cell activation, mononuclear cell differentiation, and positive regulation of lymphocyte activation were associated with the biological processes of these 53 TLS-related genes (Fig. 2a). Additionally, the analysis of KEGG revealed that the 53 TLS-related genes were involved in hematopoietic cell lineage, NF-kappa B signaling pathway, cytokine-cytokine receptor interaction, B-cell receptor signaling pathway, programmed death-ligand 1 (PD-L1) expression and programmed death-1 (PD-1) checkpoint pathway in cancer, Th1 and Th2 cell differentiation, T-cell receptor signaling pathway, Th17 cell differentiation, natural killer (NK) cell-mediated cytotoxicity, and chemokine signaling pathway (Fig. 2b). These results suggested that these TLS-related genes are involved in immunomodulatory mechanisms.
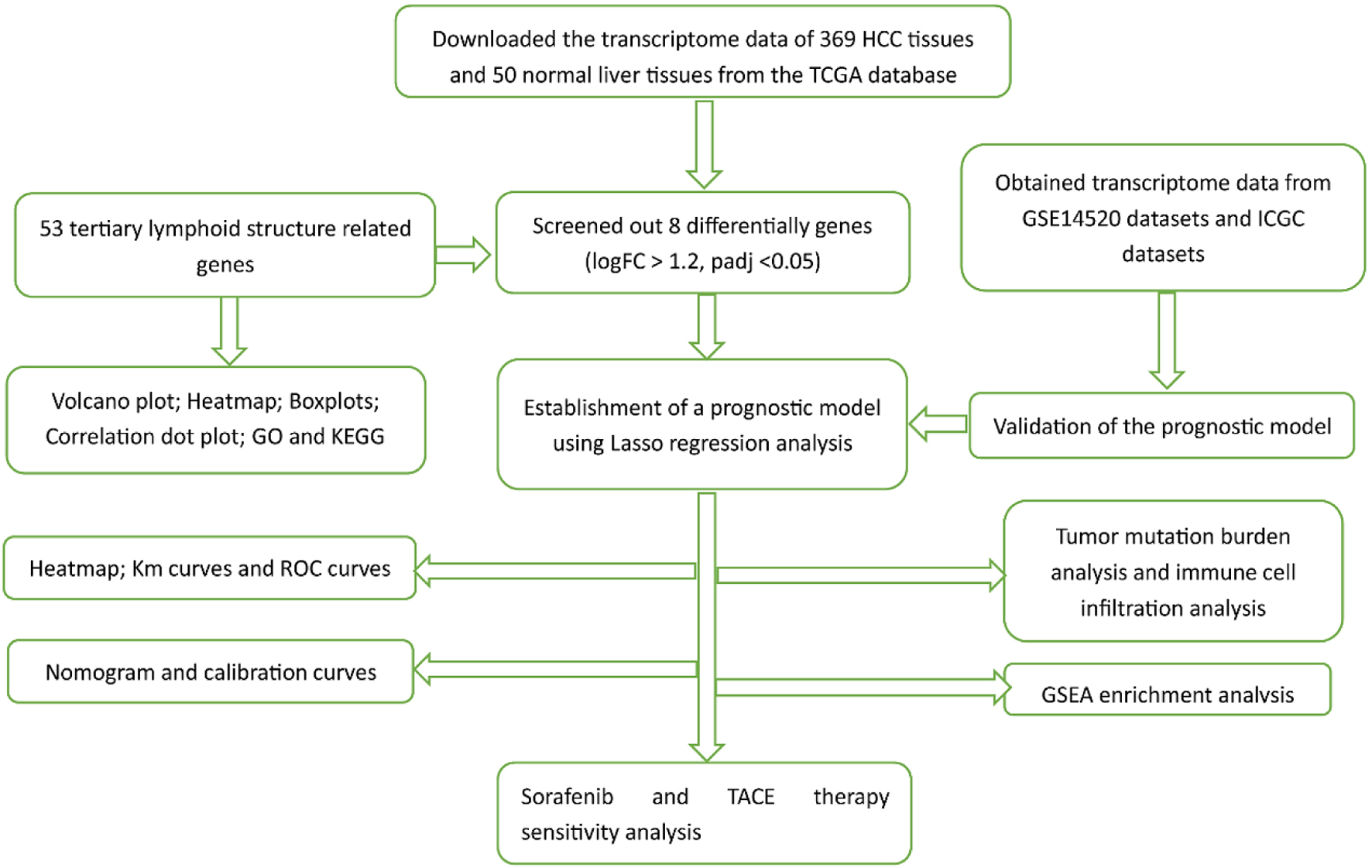 Click for large image | Figure 1. The flowchart of our study process. |
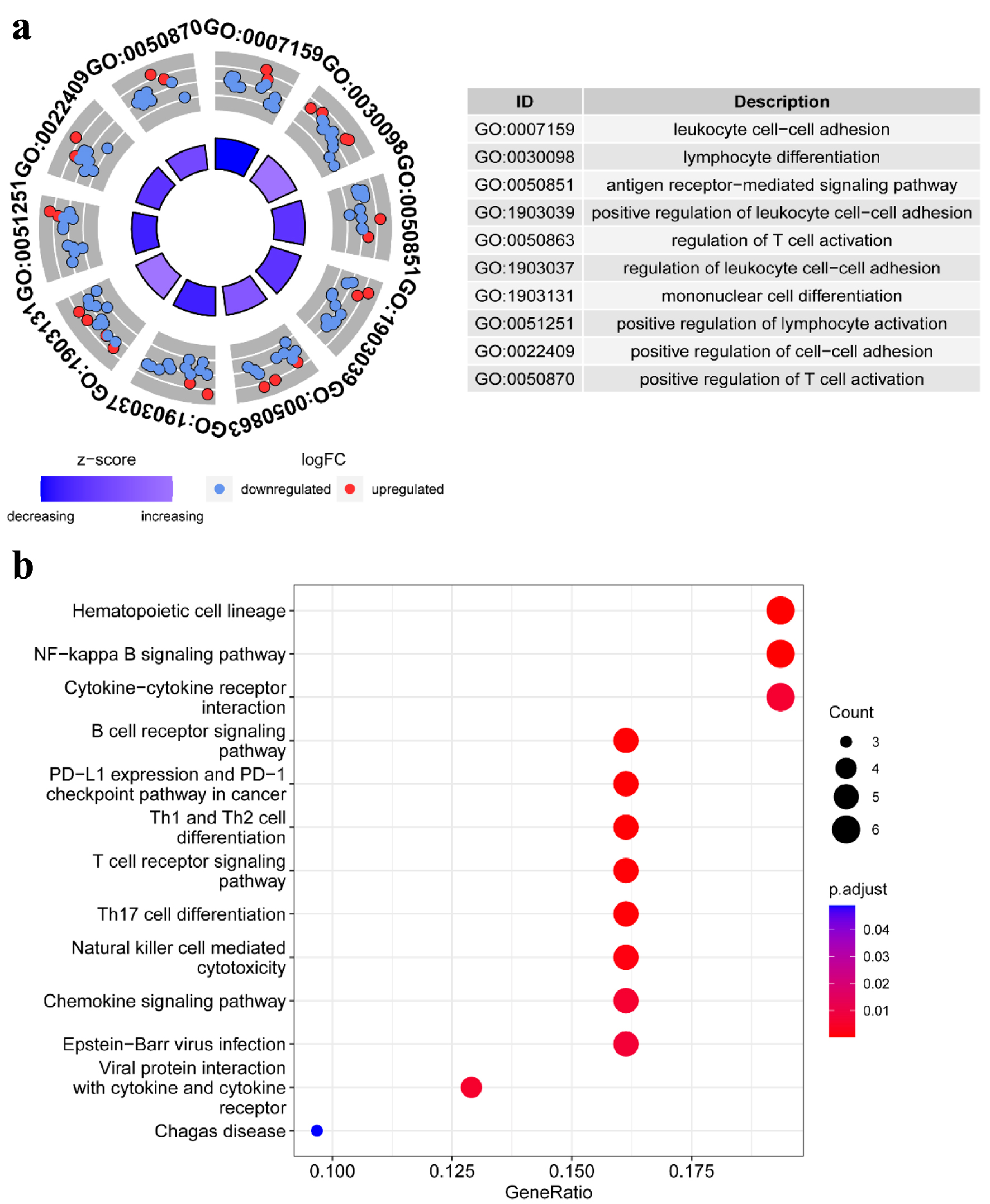 Click for large image | Figure 2. Pathway enrichment analysis of TLS-related genes in HCC patients of TCGA: (a) the enriched item in the gene ontology (GO) analysis; (b) the enriched item in the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. TLS: tertiary lymphoid structures. |
Identification of TLS-related genes that are differentially expressed in HCC
In a previous study, we obtained 53 TLS-associated genes [14]. By using the TCGA database to analyze, a more detailed gene signature, which included eight differently expressed genes between HCC tissues (n = 369) and normal tissues (n = 50), was obtained based on the cut-off criteria of |log2 FC | > 1.2 and false discovery rate (FDR) < 0.05 from 53 TLS-related genes using the R package “DESeq2”. Seven TLS-related genes were significantly downregulated in HCC tissues, according to volcano plots, heatmaps, and boxplots, whereas one TLS-related gene was elevated (Fig. 3a-c). Moreover, we investigated the correlation between the expression of different genes, which revealed strong associations (Fig. 3d).
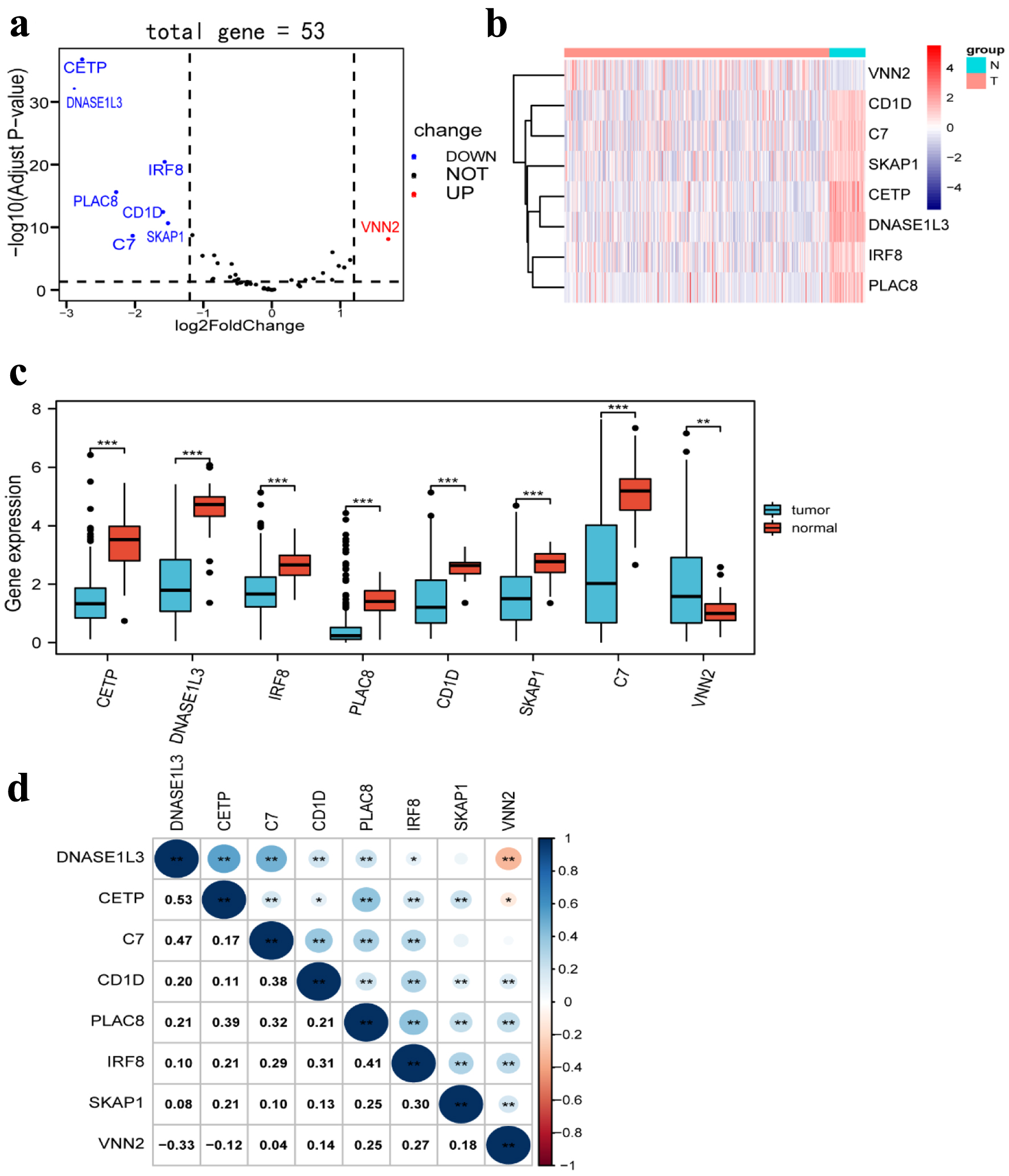 Click for large image | Figure 3. Differentially expressed TLS-related genes between HCC tissues and normal tissues. (a) Volcano plot indicates TLS-related genes, with red dots indicating high expression and blue dots indicating low expression. (b) Heatmap of differentially expressed TLS-related genes, with red indicating high expression, blue indicating low expression, n representing normal tissues, and t representing tumor tissues. (c) Boxplots of differentially expressed TLS-related genes, with blue boxes representing tumor groups and red boxes representing normal groups. (d) The correlation of TLS-related genes with each other. TLS: tertiary lymphoid structures. |
Construction of prognostic model of TLS-related genes in HCC
For all cancer samples in HCC, we created a prognostic model of TLS-related genes using LASSO regression analysis. The TLS score formula consisted of six genes: TLS score = Expression (CETP) × 0.068656 + Expression (DNASE1L3) × (-0.264498) + Expression (PLAC8) × 0.149993 + Expression (SKAP1) × (-0.137136) + Expression(C7) × (-0.026918) + Expression(VNN2) × 0.094023 (Fig. 4a, b). We classified the 363 (six patients were excluded for lack of clinical data) patients into high-TLS score (n = 181) and low-TLS score (n = 182) groups based on the median TLS score to show the prognostic model’s ability to predict outcomes (Fig. 5a). We found the high-TLS score group had longer survival times and lower mortality rates than the low-TLS score group. For HCC patients, lower scores were linked to a worse prognosis (Fig. 5b). The KM curves for OS showed that patients in the low-TLS score group had a worse prognosis (Fig. 5c). As was shown in Figure 5e, the low-TLS score group had substantially expressed levels of CETP, DNASE1L3, PLAC8, C7, and SKAP1 while having low levels of VNN2. The prognostic prediction of OS was also revealed by time-dependent ROC analysis to be 0.751 at 1 year, 0.732 at 3 years, and 0.713 at 5 years (Fig. 5d). Moreover, we assessed the association between clinical characteristics and the expression of TLS-related genes. The findings revealed that the HCC T stage and grade were inversely correlated with DNASE1L3 expression (Fig. 4c, d). The HCC T stage was likewise adversely correlated with the expression of C7 (Fig. 4e). The TLS score, however, was found to be significantly correlated with tumor T stage and grade (Fig. 4f, g). Above all, the results supported the possibility that the TLS-related gene signature in our model could be valuable for predicting HCC prognosis, and the model we developed has remarkable prognosis prediction accuracy.
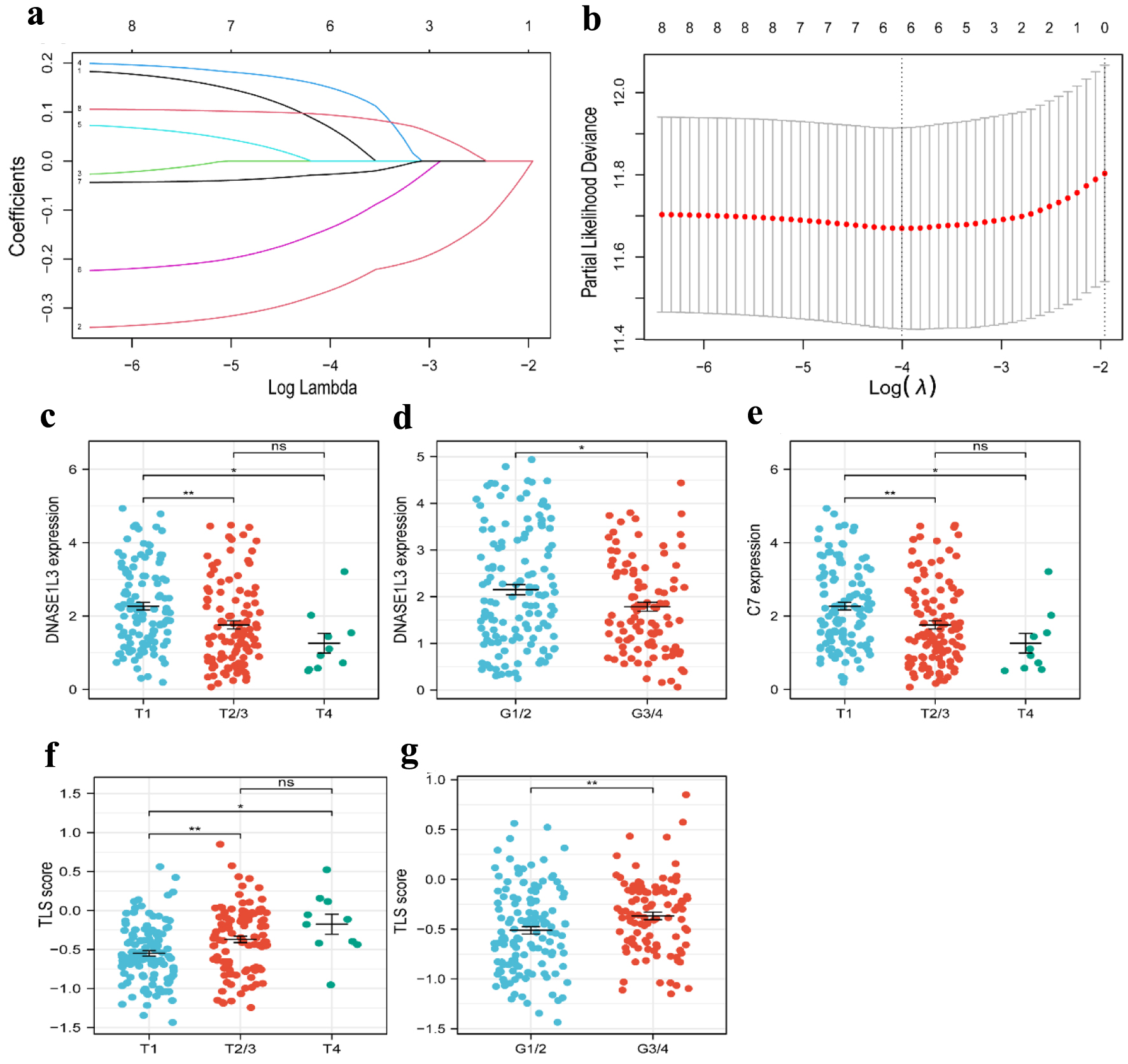 Click for large image | Figure 4. Construction of a risk prognostic model based on TLS-related genes in the TCGA cohort. (a) Cross-validation for tuning the parameter selection in the LASSO regression. (b) LASSO regression of the 6 OS-related genes. (c, d) The dot plots of T stage (c) and tumor grade (d) with DNASE1L3 expression. (e) The dot plots of T stage with C7 expression. (f, g) The dot plots of T stage (f) and grade (g) with risk score. TLS: tertiary lymphoid structures; LASSO: least absolute shrinkage and selection operator. |
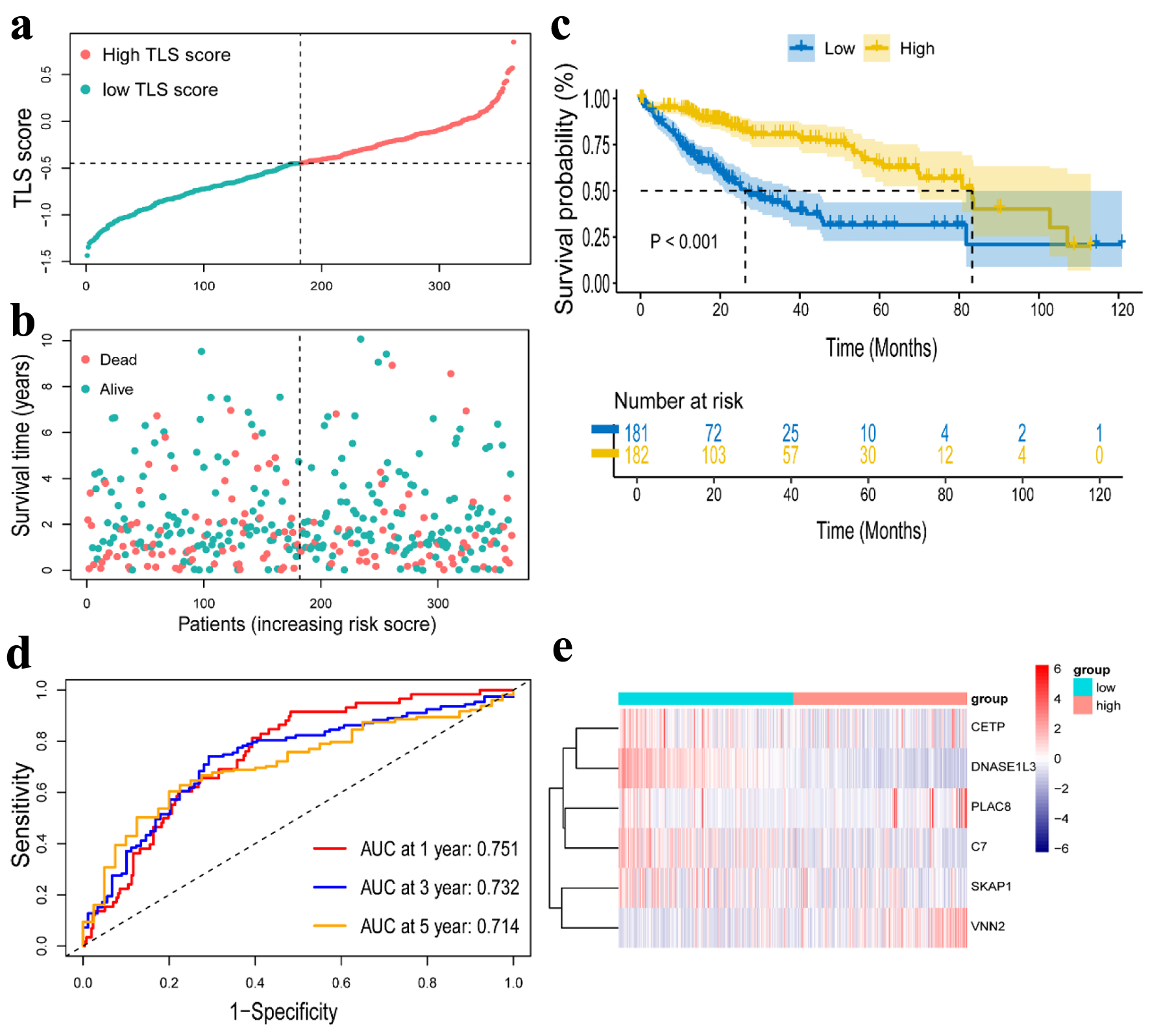 Click for large image | Figure 5. Construction of a risk prognostic model in TCGA cohort. (a) The patients were equally divided into two groups according to the threshold of the median TLS score. Blue represents the low TLS score group. Red represents the high TLS score group. (b) Survival status of patients with HCC in high and low risk groups. Blue represents survival. Red represents death. (c) Kaplan Meier curves showing the overall survival of patients in the high TLS score and low TLS score groups. (d) The predictive efficiency of the TLS score was verified by the ROC curve. (e) Heatmap showing the expression of the six TLS-related genes. Blue represents the low TLS score group. Red represents the high TLS score group. TLS: tertiary lymphoid structures. |
Verification of the predictive efficacy of TLS signature on HCC patients using the GSE14520 and ICGC dataset
We further verified the prognosis prediction capability of the TLS signature in the GSE14520 dataset. According to the same formula used to calculate TLS score in the TCGA cohort, the dataset’s 221 patients were divided into low-TLS score (n = 110) and high-TLS score (n = 111) groups (Fig. 6a, b), and comparative investigations were conducted on these two groups. While VNN2 was substantially expressed in the high-TLS score group, CETP, DNASE1L3, PLAC8, C7, and SKAP1 were highly expressed in the low-TLS score group (Fig. 6e). The high-TLS score group had an obviously longer OS than the low-TLS score group, as shown in Figure 6c. The findings in the GEO cohort were consistent with those in the TCGA cohort. Additional time-dependent ROC analysis was performed on the dataset, and the results showed that OS’s prognosis accuracy was 0.599 at 1 year, 0.635 at 3 years, and 0.698 at 5 years (Fig. 6d). The same validation was taken in the ICGC cohort, and the OS outcome was similar to the GSE 14520 cohort and the TCGA cohort (Supplementary Material 1C, www.wjon.org). Due to the short survival time of patients in the ICGC cohort, we only present the AUC for 1, 2 and 3 years (Supplementary Material 1D, www.wjon.org).
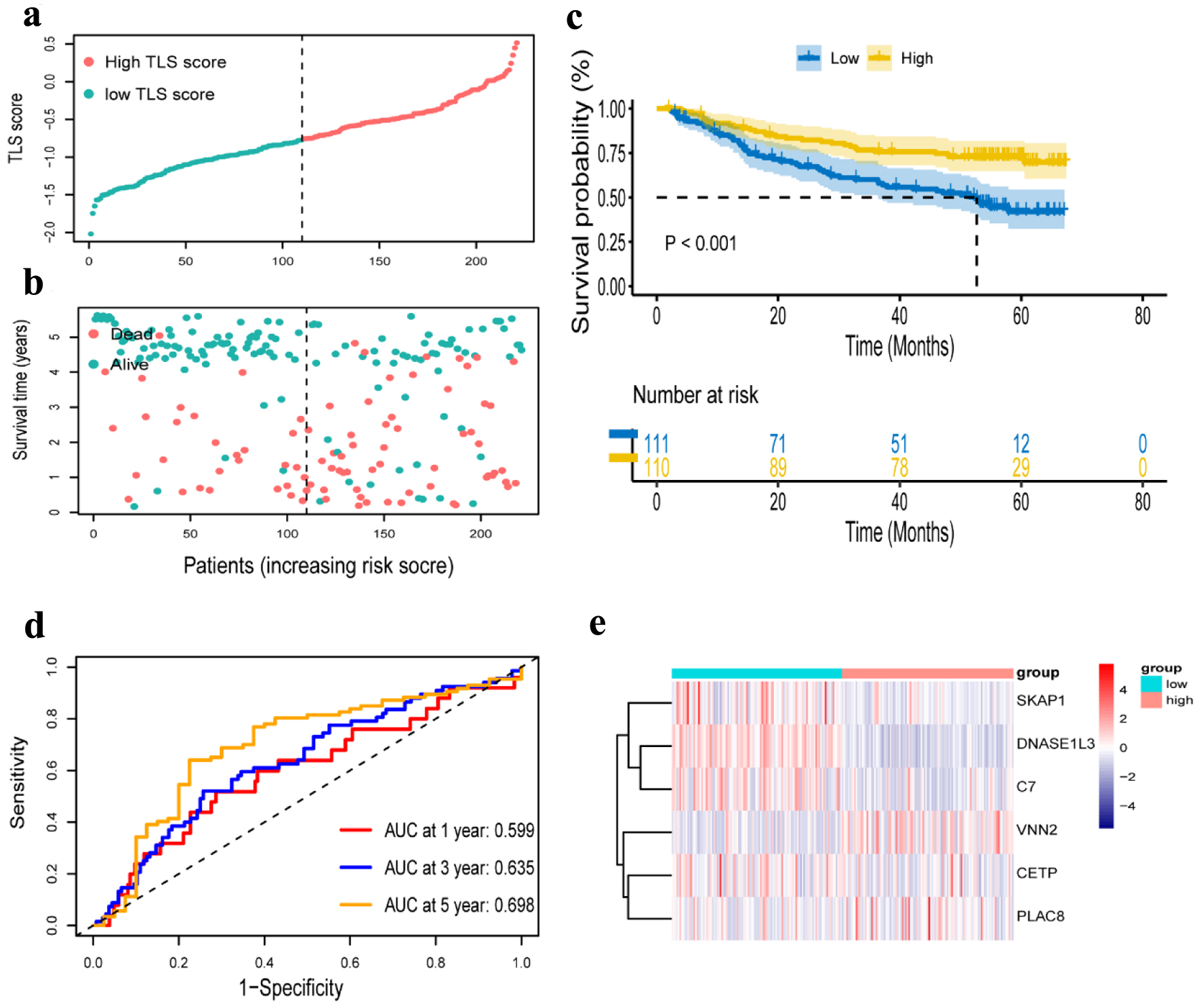 Click for large image | Figure 6. Verification of a risk prognostic model in GSE14520 cohort. (a) The patients were equally divided into two groups according to the threshold of the median risk score. Blue represents the low TLS score group. Red represents the high TLS score group. (b) Survival status of patients with HCC in high and low risk groups. Blue represents survival. Red represents death. (c) Kaplan-Meier curves showing the overall survival of patients in the high TLS score and low TLS score groups. (d) The predictive efficiency of the risk score was verified by the ROC curve. (e) Heatmap showing the expression of the six TLS-related genes. Blue represents the low TLS score group. Red represents the high TLS score group. TLS: tertiary lymphoid structures. |
Independent prognostic analysis of TLS score and establishment of nomogram
We conducted univariate and multivariate Cox regression analysis to investigate whether the TLS score and clinical features could serve as separate prognostic variables. M, stage, and TLS score were found to be closely linked with the OS of HCC patients by univariate Cox analysis (Fig. 7a). The TLS score could act individually as a prognostic predictor, according to multivariate Cox prognostic analysis (P < 0.0001) (Fig. 7b). Additionally, in order to give clinicians a more accurate quantitative way to predict the prognosis of patients with HCC, we created a nomogram that combines gender, stage, T, N, M, and TLS score. The nomogram demonstrated that the TLS score was a significant prognostic predictor among diverse clinical characteristics (Fig. 8a). Moreover, we established calibration curves, which demonstrated that the nomogram matched the real survival of HCC patients with good accuracy (Fig. 8b-d). Our prognostic model also had a higher AUC value (AUC = 0.798, Fig. 8e). Based on these findings, we hypothesized that our model would function as a trustworthy prognostic biomarker.
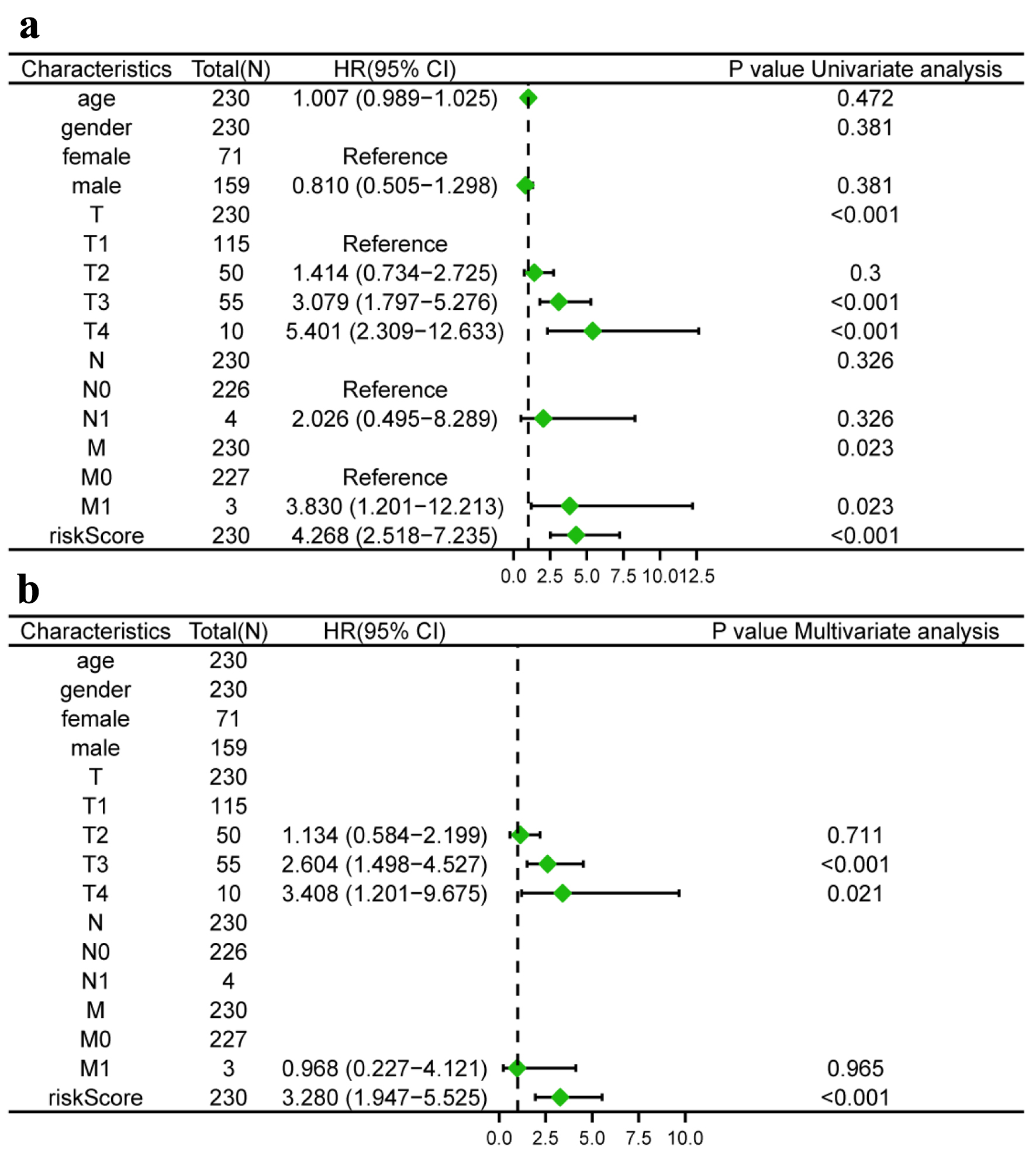 Click for large image | Figure 7. Risk model independent prognostic analysis. (a) Univariate independent prognosis Cox regression analysis of TLS score and indicated clinical characteristics. (b) Multivariate independent prognosis Cox regression analysis of TLS score and indicated clinical characteristics. TLS: tertiary lymphoid structures. |
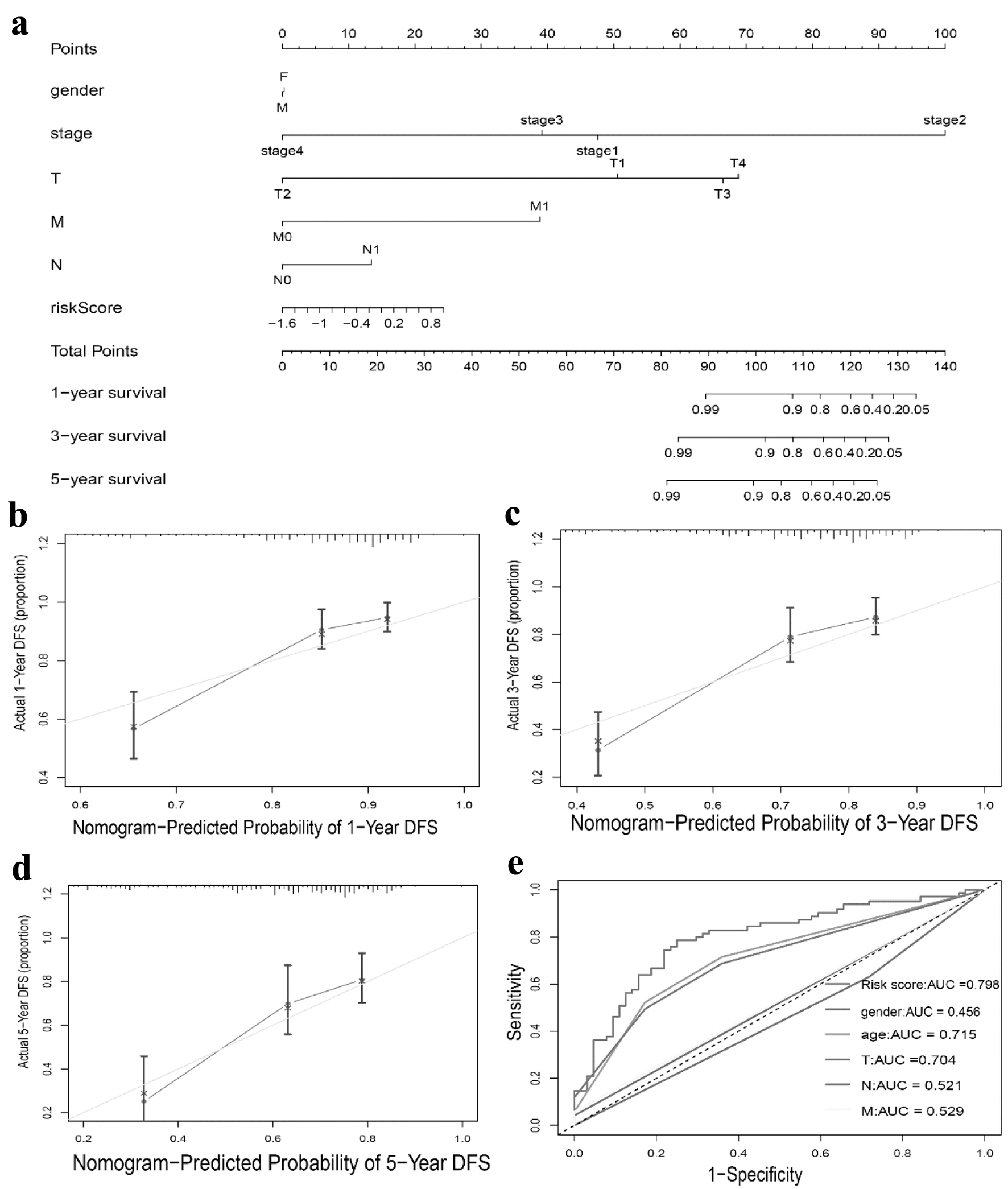 Click for large image | Figure 8. Nomogram to predict survival probability of hepatocellular carcinoma patients. (a) Nomogram combining TLS score with pathologic features. (b-d) Calibration plots for predicting 1 -, 3 -, 5-year OS of patients. (e) ROC curves for prediction of survival by the TLS score and other variables (age, gender, T stage, N stage, M stage). TLS: tertiary lymphoid structures. |
GSEA enrichment analysis
To explore the biological processes of our TLS signature, we performed a GSEA enrichment analysis after differential analysis of the high- and low-TLS score groups. Results from the high-TLS score group revealed that cornification, keratinization, ncRNA 3-end processing, snRNA 3-end processing, and snRNA processing were among the significantly enriched pathways (Supplementary Material 2A, www.wjon.org). The primary enhanced pathways in the low-TLS score group, according to our data, were the cellular amino acid catabolic process, cellular response to copper ions, fatty acid catabolic process, monocarboxylic acid catabolic process, and organic acid catabolic process (Supplementary Material 2B, www.wjon.org).
Genetic alterations landscape of TLS
Genetic alteration analysis revealed significant differences between the low- and high-TLS score groups in the mutation rates of the top 30 most significantly altered genes, as shown in Figure 9a, b. We estimated the TMB for each patient in the TCGA cohort and performed a correlation study between the TMB and TLS scores. The results revealed no significant link between the two (r = 0.186, P < 0.001, Supplementary Material 3A, www.wjon.org). After dividing the patients into groups with high and low TMB using the median TMB as the cut-off, we combined this cut-off with the TLS score level to produce four more specific subgroups (TMB_L&TLS_L, TMB_L&TLS_H, TMB_H&TLS_L, and TMB_H&TLS_H). Additionally, we conducted a survival study for these four subgroups, and the results revealed that patients in the TMB_H&TLS_H group had higher survival rates (Fig. 9c).
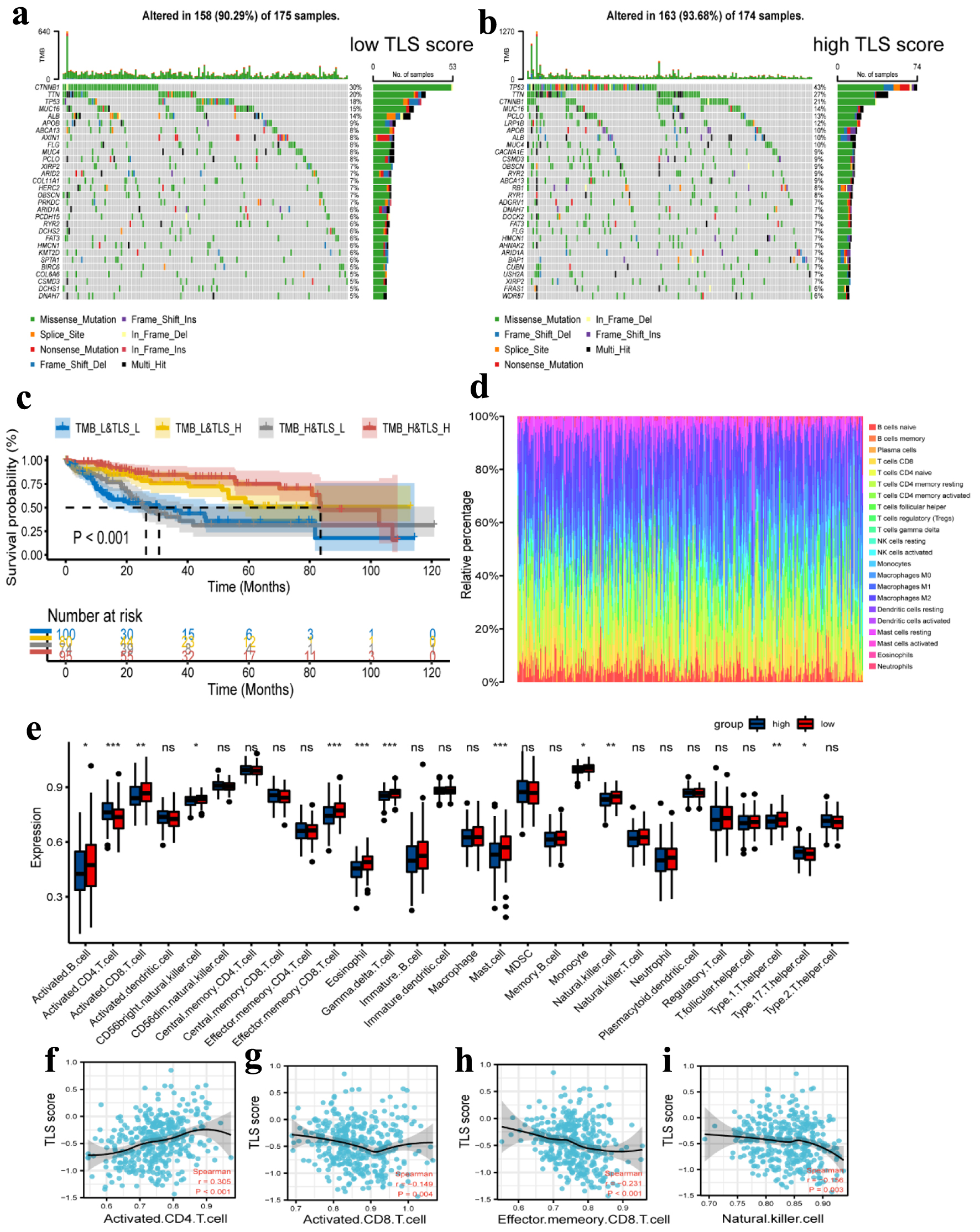 Click for large image | Figure 9. Tumor mutation analysis and immune cell infiltration analysis. (a, b) Waterfall plots of the mutated genes in the HCC patients from low TLS score (a) and high TLS score (b) subgroups. The mutation types of genes were marked by different colors. (c) Kaplan Meier curves showing the overall survival of patients in the TMB_L&TLS_L, TMB_L&TLS_H, TMB_H&TLS_L, and TMB_H&TLS_H groups. (d) Relative proportion of immune cell infiltration in high TLS score and low TLS score group. (e) Different expression of immune cell infiltration in high TLS score and low TLS score group. Red represents the high TLS score group. Blue represents the low TLS score group. (f-i) Correlation between activated CD4 T cell (f), activated CD8 T cell (g), effector memory CD8 T cell (h), and natural killer cell (i) and TLS score in HCC. TLS: tertiary lymphoid structures. |
Immune cell infiltration profile of TLS
Additionally, the CIBERSORT method was used to calculate the fraction of 22 subtypes of immune cells in HCC patients, and a cut-off value of P-value 0.05 was used (Fig. 9d). In comparison to the low-risk group, the high-TLS score group had lower levels of activated B cells, CD8 T cells, CD56bright NK cells, effector memory CD8 T cells, eosinophils, gamma delta T cells, mast cells, monocytes, NK cells, and type 1 T helper cells. However, only type 17 T helper cells and the infiltration level of activated CD4 T cells were higher in the high-TLS score group compared to the low-TLS score group (Fig. 9e). In addition, we examined the relationship between the TLS score and the four immune cells. The results showed that only activated CD4 T cells linked positively with TLS score, but activated CD8 T cells, effector memory CD8 T cells, and NK cells all correlated negatively with TLS score (Fig. 9f-i). Other correlation plots of relative immune cells are shown in Supplementary Material 3 (www.wjon.org). Together, these findings provide us reasons to believe that the activated CD4 T cell may be crucial to the development and subsequent function of TLS.
Prediction of TLS score in HCC therapy
To confirm the prediction of TLS score on the response to HCC treatment, we studied the responsiveness of patients to sorafenib and TACE treatment in different TLS score groups. According to Figure 10a, the high-TLS score group had a higher probability of responding to sorafenib than the low-TLS score group. In order to predict the responsiveness to sorafenib, we also created an ROC curve, and the AUC value was 0.887 (Fig. 10b). These findings suggested that sorafenib treatment would be effective for HCC patients with a higher TLS score. In the sorafenib cohort, we examined the expression of TLS-related genes and discovered that CETP and PLAC8 were substantially expressed in the responder group, whereas DNASE1L3, SKAP1, C7, and VNN2 were highly expressed in the non-responder group (Fig. 10c). In addition, we assessed the association between TLS-related genes and responsiveness to transcatheter arterial chemoembolization (TACE), a common treatment for HCC patients at stage BCLC A or B [20]. Similarly, we performed a comparative study in the TACE treatment cohort. It was interesting to see that patients who responded to sorafenib had decreased responsiveness to TACE (Fig. 10d), and the AUC value of the TLS score in predicting responsiveness to TACE was 0.75 (Fig. 10e). Only four genes, DNASE1L3, SKAP1, C7, and VNN2, were found to be differentially expressed in the TACE cohort, as depicted in Figure 10f. Furthermore, and in stark contrast to the findings of the sorafenib cohort, these four genes were more highly expressed in the responder group than in the non-responder group. In order to further investigate the relationship between TLS and immunotherapy, we assessed the expression of immune checkpoint-related genes in different TLS score groups. The results revealed that CTLA4, HAVCR2, PDCD1, and TIGIT were all more highly expressed in the high TLS score group (Fig. 10g), suggesting that patients with high TLS scores may benefit from immunotherapy. Otherwise, we used the TIDE website to predict how sensitive HCC patients would be to immunotherapy, and the findings demonstrated that those with higher TLS scores had better sensitivity to anti-PD-1 and anti-CTLA4 treatment (Fig. 10h).
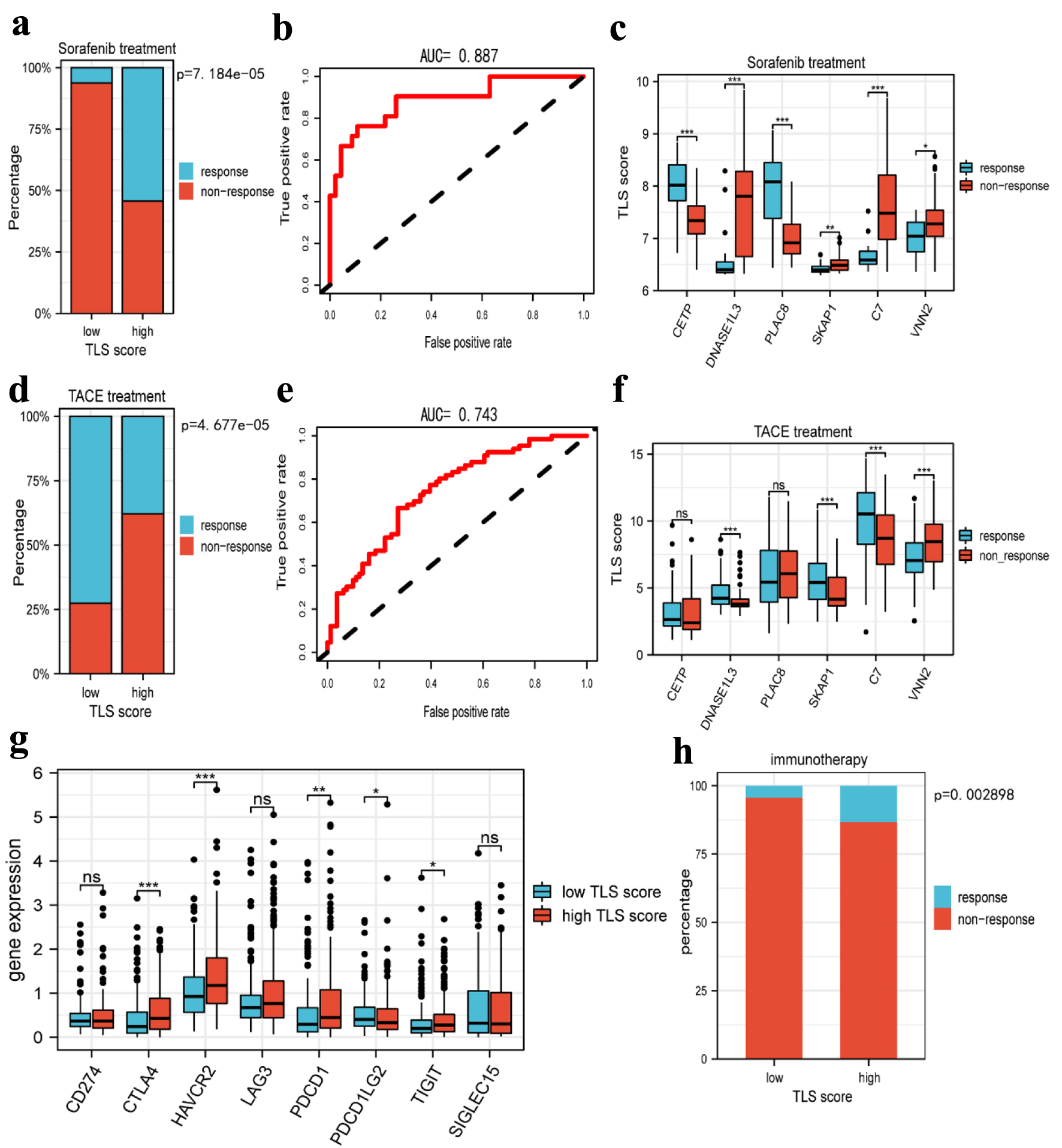 Click for large image | Figure 10. Prediction of tertiary lymphoid structures related TLS score in HCC therapy. (a) The histogram showing that the ratio of response to sorafenib between low and high TLS score group. (b) The AUC value of TLS score in predicting the efficiency of sorafenib in HCC patients. (c) The boxplot showing the expression of TLS-related genes in the responders and non-responders to sorafenib. (d) The histogram showing that the ratio of response to TACE between low and high TLS score group. (e) The AUC value of TLS score in predicting the efficiency of TACE in HCC patients. (f) The boxplot showing the expression of TLS-related genes in responders and non-responders to TACE. (g) Boxplots indicate the expression of immune checkpoints genes in high and low TLS score group. (h) The sensitivity of the two subtypes to immunotherapy in TCGA cohorts was predicted using the TIDE website. TLS: tertiary lymphoid structures; TACE: transcatheter arterial chemoembolization; TIDE: Tumor Immune Dysfunction and Exclusion. |
| Discussion | ▴Top |
HCC is a highly malignant tumor characterized by high morbidity, metastasis, and recurrence rates, significantly threatening the lives of patients [1-3]. TLS are immune structures formed by a variety of immune-related cells in response to antigenic stimuli, such as chronic inflammation and tumors [6, 11, 14]. Numerous studies have demonstrated the protective effect of third lymphatic structures on a variety of tumors, including melanoma, lung cancer and HCC [14, 21, 22]. However, previous studies primarily focused on the pathological level to measure the presence or absence of TLS and neglected to investigate its prognostic significance in different diseases. Therefore, we developed and validated the predictive efficacy of this six-gene model for the prognosis and treatment response of HCC patients using the LASSO regression algorithm. In our research, through the analysis of differentially expressed TLS-related genes, we identified a TLS prognostic model that could serve as a potential favorable factor for HCC. Compared to the high-TLS score group, patients in the low-TLS score group exhibited poorer prognoses, more advanced disease stages, higher TMBs, and elevated levels of immune cell infiltration. Moreover, subsequent analyses revealed that HCC patients in the low-TLS score group exhibited poorer responses to sorafenib, while demonstrating better responses to TACE treatment. Therefore, our TLS prognostic model can play an important role in guiding clinical decisions and treatment options for HCC patients.
In recent years, the beneficial role of TLS has been extensively studied, and numerous researchers have developed prognostic signatures based on TLS measurements at the pathological tissue level [21, 23-25]. However, the evaluation of TLS through staining markers presents several limitations. Our findings utilizing bulk-RNA sequencing technology offer more accurate predictions of prognostic risks in HCC patients and allow more results to be obtained. Additionally, the original TLS gene signature we used mainly represents B and T cells within TLS and is strongly associated with B-cell markers. Thus, we believe that our six-gene signature, representing the TLS-related gene expression signature, may exhibit superior predictive performance in HCC compared to other existing prognostic signatures.
In this study, we re-analyzed the differential expression of the TLS gene set between HCC tissues and normal tissues and finally screened eight genes with significant differences in expression, of which one gene was overexpressed and seven were underexpressed. Utilizing these eight differentially expressed genes, we developed a six-gene prognostic model through LASSO regression analysis, and subsequently validated the model via univariate and multivariate regression analyses. Of the six genes, only VNN2 was highly expressed in the high-TLS score group, whereas the other five genes were all expressed at low levels. CETP plays a role in plasma cholesterol transport and can reduce plasma high-density lipoprotein (HDL) cholesterol levels. A recent study has found that CETP expression in macrophages can regulate mitochondrial function, enhancing cellular antioxidant capacity to achieve anti-inflammatory effects [26]. DNASE1L3 is a member of the deoxyribonuclease I family, which encodes proteins that digest DNA in chromatin. DNASE1L3 has been identified as a prognostic biomarker for HCC, inhibiting tumor growth through the suppression of the PI3K/AKT signaling pathway [27]. PLAC8 is a lysosomal protein that localizes to the inner surface of the plasma membrane and interacts with other cellular components to regulate autophagy and cell cycle arrest. Chen et al demonstrated that high expression of PLAC8 correlates with a worse prognosis and drug resistance in breast cancer patients [28]. SKAP1 encodes a T-cell adaptor protein, a class of intracellular molecules with modular domains capable of recruiting additional proteins without intrinsic enzymatic activity. SKAP1 plays an important role in the regulation of PLK1 kinase activity, thereby regulating T-cell function [29]. C7 is a component of the complement system. It participates in the formation of the membrane attack complex (MAC). Its expression level was a potential prognostic predictor for both disease progression and death in patients with NSCLC [30]. VNN2 belongs to the Vanin protein family, sharing extensive sequence similarity with each other and with biotinidase. Previous studies have shown that VNN2 can be used as a prognostic marker for HCC [31]. To our knowledge, this is the first TLS-related predictive model for HCC developed using these six genes. We repeated the analysis in the GSE14520 and ICGC cohorts using the same TLS score calculation formula to further validate the applicability of our TLS model. KM analysis revealed that the high-TLS score group in both cohorts had a longer OS, consistent with the findings from the TCGA cohort. ROC curves also demonstrated that the TLS score model exhibited exceptionally high accuracy in two separate cohorts. Additionally, the calibration curve, univariate Cox analysis, and multivariate Cox analysis results showed that the TLS score was related to HCC clinical traits. In conclusion, these results indicated that the TLS score could function as an independent prognostic biomarker for HCC patients.
To elucidate the underlying biological mechanisms of the TLS signature, we initially performed functional enrichment analysis in HCC patients. Specifically, PD-L1 expression and the PD-1 checkpoint pathway in cancer were found to be associated with these TLS signature genes. This finding suggests that TLS may be more susceptible to the effectiveness of immunotherapy. Subsequently, we conducted GSEA analysis between high- and low-TLS score groups. The results indicated that the majority of the related pathways in the low-TLS score group were metabolic pathways, including pathways for amino acids, fatty acids, the catabolic process of organic acids, and cellular responses to copper ions. Based on these enriched pathways, novel medications could be developed to specifically target certain metabolic processes.
A recent study indicated that the HCC phenotype appears to be closely associated with particular gene mutations, tumor subgroups, and oncogenic pathways [32]. Analysis of the gene mutation profile in HCC revealed that patients with low TLS scores exhibited higher mutation frequencies in CTNNB1. A recent study confirmed that TMB values were higher in HCC patients with CTNNB1 mutations, who also had worse prognoses and clinicopathology [33]. Overall, this result validated our hypothesis that gene mutation events are associated with poorer prognosis in the low-TLS score group, consistent with our research. Otherwise, the TME is a complex network formed by the interaction of multiple cellular components, including malignant cells, endothelial cells, stromal cells, and immune cells [34]. And considering TLS as an immune structure, we investigated the immune infiltration level of TLS signature genes in HCC patients with different TLS scores. Immuno-infiltration analysis results indicated that the TLS score was associated with CD4T, CD8T, and NK cells. As we all know, all of these cells play a role in resisting HCC progression and exerting anti-tumor effects [35], suggesting that the TLS score can reflect the altered immune function during the progression of HCC.
As a multi-kinase inhibitor of the oral vascular endothelial growth factor receptor [36], the relationship between sorafenib and TLS remains unclear. Our further study found that the TLS score was a reliable predictor of response to sorafenib in HCC patients. Patients with a higher TLS score exhibited better sensitivity to sorafenib, whereas those with a lower TLS score showed resistance to sorafenib. It remains unclear whether TLS or other biological effects influence the responsiveness of HCC patients to sorafenib, and the mechanism involved requires further study. Previous studies have demonstrated the predictive value of TLS on immunotherapy response [6], and to validate the utility of our TLS score, we examined the expression of immune checkpoint-related genes in the high- and low-TLS score groups. As expected, our findings indicated that HCC patients with high TLS scores exhibited significantly elevated expression of immune checkpoint-related genes, such as CTLA4, HAVCR2, PDCD1, and TIGIT, indicating a greater likelihood of benefiting from immunotherapy. In order to further explore the potential significance of the TLS score in immunotherapy prediction, we used the TIDE website to forecast the sensitivity to immunotherapy. The results showed that the group with a high TLS score had a greater response rate. These findings could potentially lead to new therapeutic targets for the clinical treatment of HCC.
The ultimate goal of all medical research is its clinical application. Our study successfully developed a novel prognostic model related to TLS and validated its reliability and predictiveness for some treatments. However, there are still many shortcomings in our study that need further improvement in our future studies. First, our study is basically an analysis of data obtained by bulk-RNA sequencing, and further in vivo and in vitro experiments are needed to validate it in the future. Second, many potential mechanisms for the six TLS genes in our results need to be further explored in our subsequent studies. Finally, more clinical studies are required to verify the advantages of high TLS scores for the therapy recommendations for HCC patients.
Conclusion
In conclusion, we developed a novel TLS-related prognostic model for HCC in the TCGA cohort and the GSE14520 cohort and the LIRI-JP cohort. Our results showed that patients with high TLS scores had longer survival compared to those with low TLS scores and that patients with high TLS scores had higher responsiveness to sorafenib and immunotherapy and lower responsiveness to TACE.
| Supplementary Material | ▴Top |
Suppl 1. Verification of a risk prognostic model in ICGC cohort. (A) The patients were equally divided into two groups according to the threshold of the median risk score. Blue represents the low TLS score group. Red represents the high TLS score group. (B) Survival status of patients with HCC in high and low risk groups. Blue represents survival. Red represents death. (C) Kaplan-Meier curves showing the overall survival of patients in the high TLS score and low TLS score groups. (D) The predictive efficiency of the risk score was verified by the ROC curve. (E) Heatmap showing the expression of the six TLS-related genes. Blue represents the low TLS score group. Red represents the high TLS score group. TLS: tertiary lymphoid structures.
Suppl 2. GSEA analysis. (A) GSEA enrichment analysis results of GO database in high TLS score group. (B) GSEA enrichment analysis results of GO database in low TLS score group. GO: Gene Ontology; TLS: tertiary lymphoid structures.
Suppl 3. The correlation of immune infiltration cells with TLS risk score. (A) The correlation of TLS score with TMB. (B-I) The correlation of TLS risk score with activated B cell (B), CD56bright natural killer cell (C), gamma delta cell (D), eosinophil (E), mast cell (F), monocyte (G), type1 T helper cell (H), and type17 T helper cell (I). TLS: tertiary lymphoid structures; TMB: tumor mutation burden.
Acknowledgments
None to declare.
Financial Disclosure
This work was supported by the Key Science and Technology Projects of Henan Province (No. 232102311048).
Conflict of Interest
The authors declare that they have no conflict of interest.
Informed Consent
All subjects provided written informed consent.
Author Contributions
Yin Liu and Chao Bo Li wrote the main article as well as drew the main graphics. Yun Peng Zhai, Ding Yang Li and Shao Kang Zhang were responsible for optimizing the graphics and correcting the errors in statement. Ruo Peng Liang and Zhi Qiang Gao were responsible for the overall instruction of the article and the optimization of the graphics.
Data Availability
The datasets analyzed during the current study are available in the TCGA (https://portal.gdc.cancer.gov/projects, TCGA-LIHC Project) and GEO repository (https://www.ncbi.nlm.nih.gov/geo/, accession number GSE14520, 104580, 109211) and ICGC portal (https://icgc.org/).
| References | ▴Top |
- Singal AG, Lampertico P, Nahon P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J Hepatol. 2020;72(2):250-261.
doi pubmed pmc - Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6.
doi pubmed - Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589-604.
doi pubmed pmc - Gabutti A, Bhoori S, Cascella T, Bongini M. Hepatocellular carcinoma recurrence after liver transplantation. Oncology (Williston Park). 2020;34(3):692516.
pubmed - Tabrizian P, Jibara G, Shrager B, Schwartz M, Roayaie S. Recurrence of hepatocellular cancer after resection: patterns, treatments, and prognosis. Ann Surg. 2015;261(5):947-955.
doi pubmed - Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19(6):307-325.
doi pubmed - Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, Lepelley A, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med. 2014;189(7):832-844.
doi pubmed - Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S, de Chaisemartin L, et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res. 2014;74(3):705-715.
doi pubmed - Munoz-Erazo L, Rhodes JL, Marion VC, Kemp RA. Tertiary lymphoid structures in cancer - considerations for patient prognosis. Cell Mol Immunol. 2020;17(6):570-575.
doi pubmed pmc - Martinet L, Filleron T, Le Guellec S, Rochaix P, Garrido I, Girard JP. High endothelial venule blood vessels for tumor-infiltrating lymphocytes are associated with lymphotoxin beta-producing dendritic cells in human breast cancer. J Immunol. 2013;191(4):2001-2008.
doi pubmed - Sautes-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman WH, Dieu-Nosjean MC. Tertiary lymphoid structures in cancers: prognostic value, regulation, and manipulation for therapeutic intervention. Front Immunol. 2016;7:407.
doi pubmed pmc - Cottrell TR, Thompson ED, Forde PM, Stein JE, Duffield AS, Anagnostou V, Rekhtman N, et al. Pathologic features of response to neoadjuvant anti-PD-1 in resected non-small-cell lung carcinoma: a proposal for quantitative immune-related pathologic response criteria (irPRC). Ann Oncol. 2018;29(8):1853-1860.
doi pubmed pmc - Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, Kiialainen A, et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med. 2018;24(7):994-1004.
doi pubmed pmc - Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561-565.
doi pubmed - Yan C, Niu Y, Li F, Zhao W, Ma L. System analysis based on the pyroptosis-related genes identifies GSDMC as a novel therapy target for pancreatic adenocarcinoma. J Transl Med. 2022;20(1):455.
doi pubmed pmc - Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545-15550.
doi pubmed pmc - Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol Biol. 2018;1711:243-259.
doi pubmed pmc - Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, Porta-Pardo E, et al. The immune landscape of cancer. Immunity. 2018;48(4):812-830.e814.
doi pubmed pmc - Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24(10):1550-1558.
doi pubmed pmc - Marrero JA, Kulik LM, Sirlin CB, Zhu AX, Finn RS, Abecassis MM, Roberts LR, et al. Diagnosis, staging, and management of hepatocellular carcinoma: 2018 practice guidance by the American Association for the study of liver diseases. Hepatology. 2018;68(2):723-750.
doi pubmed - Calderaro J, Petitprez F, Becht E, Laurent A, Hirsch TZ, Rousseau B, Luciani A, et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J Hepatol. 2019;70(1):58-65.
doi pubmed - Feng H, Yang F, Qiao L, Zhou K, Wang J, Zhang J, Tian T, et al. Prognostic significance of gene signature of tertiary lymphoid structures in patients with lung adenocarcinoma. Front Oncol. 2021;11:693234.
doi pubmed pmc - Li H, Wang J, Liu H, Lan T, Xu L, Wang G, Yuan K, et al. Existence of intratumoral tertiary lymphoid structures is associated with immune cells infiltration and predicts better prognosis in early-stage hepatocellular carcinoma. Aging (Albany NY). 2020;12(4):3451-3472.
doi pubmed pmc - Li H, Liu H, Fu H, Li J, Xu L, Wang G, Wu H. Peritumoral tertiary lymphoid structures correlate with protective immunity and improved prognosis in patients with hepatocellular carcinoma. Front Immunol. 2021;12:648812.
doi pubmed pmc - Jia W, Yao Q, Wang Y, Mao Z, Zhang T, Li J, Nie Y, et al. Protective effect of tertiary lymphoid structures against hepatocellular carcinoma: New findings from a genetic perspective. Front Immunol. 2022;13:1007426.
doi pubmed pmc - Dorighello GG, Assis LHP, Rentz T, Morari J, Santana MFM, Passarelli M, Ridgway ND, et al. Novel role of CETP in macrophages: reduction of mitochondrial oxidants production and modulation of cell immune-metabolic profile. Antioxidants (Basel). 2022;11(9):1734.
doi pubmed pmc - Chen QY, Li L, Suo DQ. DNase1L3 suppresses hepatocellular carcinoma growth via inhibiting complement autocrine effect. Neoplasma. 2021;68(4):683-691.
doi pubmed - Chen Y, Jia Y, Mao M, Gu Y, Xu C, Yang J, Hu W, et al. PLAC8 promotes adriamycin resistance via blocking autophagy in breast cancer. J Cell Mol Med. 2021;25(14):6948-6962.
doi pubmed pmc - Raab M, Strebhardt K, Rudd CE. Immune adaptor SKAP1 acts a scaffold for Polo-like kinase 1 (PLK1) for the optimal cell cycling of T-cells. Sci Rep. 2019;9(1):10462.
doi pubmed pmc - Ying L, Zhang F, Pan X, Chen K, Zhang N, Jin J, Wu J, et al. Complement component 7 (C7), a potential tumor suppressor, is correlated with tumor progression and prognosis. Oncotarget. 2016;7(52):86536-86546.
doi pubmed pmc - Li W, Lu J, Ma Z, Zhao J, Liu J. An integrated model based on a six-gene signature predicts overall survival in patients with hepatocellular carcinoma. Front Genet. 2019;10:1323.
doi pubmed pmc - Calderaro J, Ziol M, Paradis V, Zucman-Rossi J. Molecular and histological correlations in liver cancer. J Hepatol. 2019;71(3):616-630.
doi pubmed - Wang S, Shi H, Liu T, Li M, Zhou S, Qiu X, Wang Z, et al. Mutation profile and its correlation with clinicopathology in Chinese hepatocellular carcinoma patients. Hepatobiliary Surg Nutr. 2021;10(2):172-179.
doi pubmed pmc - Arneth B. Tumor microenvironment. Medicina (Kaunas). 2019;56(1):15.
doi pubmed pmc - El-Rebey HS, Abdou AG, Sultan MM, Ibrahim SH, Holah NS. The profile and role of tumor-infiltrating lymphocytes in hepatocellular carcinoma: an immunohistochemical study. Appl Immunohistochem Mol Morphol. 2021;29(3):188-200.
doi pubmed - Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378-390.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.