

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website http://www.wjon.org |
Case Report
Volume 8, Number 2, April 2017, pages 53-57
Gliosarcoma in Young Adults: A Rare Variant of Glioblastoma
Neha Kakkara, Jaspreet Kaura, d, Gunjesh Kumar Singha, Pragya Singhb, Fouzia Sirajc, Ajay Guptaa
aDepartment of Radiotherapy, VMMC & Safdarjung Hospital, New Delhi 110029, India
bDepartment of Pathology, VMMC & Safdarjung Hospital, New Delhi 110029, India
cNational Institute of Pathology, New Delhi 110029, India
dCorresponding Author: Jaspreet Kaur, Room No. 266, Second Floor, New OPD Block, Vardhman Mahavir Medical College & Safdarjung Hospital, New Delhi 110029, India
Manuscript accepted for publication March 29, 2017
Short title: Gliosarcoma in Young Adults
doi: https://doi.org/10.14740/wjon998w
| Abstract | ▴Top |
Gliosarcoma is a rare central nervous system (CNS) malignancy. It is characterized by classical biphasic histological pattern with both glial and sarcomatous components, often seen in fifth and sixth decade of life. They are generally located in the supratentorial region. Due to its rarity, exact treatment recommendations are not available in literature. Since it is considered as a variant of glioblastoma multiforme (GBM), it is treated with surgery followed by adjuvant radiotherapy and temozolomide-based chemotherapy. We present a series of four cases of this rare malignancy that were treated at our institute.
Keywords: Gliosarcoma; Surgery; Chemotherapy; Radiotherapy; Young adults; Outcome
| Introduction | ▴Top |
Gliosarcoma is a rare primary malignant tumor of central nervous system with reported incidence being 0.59-0.76% of all adult brain tumors. It has a biphasic morphological pattern with both glial and malignant mesenchymal components. It is considered as a variant of glioblastoma multiforme (GBM) representing only 2% of all GBM cases [1]. It usually affects fifth and sixth decade of life (average age of onset is 54 years) with slight male preponderance (males being affected twice as often as females) [2]. Therapeutic modalities are surgical resection, external beam radiotherapy and chemotherapy but prognosis remains poor in terms of survival [3]. We present here four cases of gliosarcoma who were treated at our institute (Table 1).
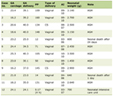 Click to view | Table 1. Patient Characteristics and Treatment Details |
| Case Reports | ▴Top |
Case 1
A 35-year-old gentleman presented in 2012 to neurosurgery outpatient department with complaints of headache and vomiting for 1 month. There was no deficit in neurocognitive functions. Non-contrast computed tomography (NCCT) of head was done which showed an approximately 6.5 × 4.4 cm sized heterogeneous lesion containing multiple necrotic areas in left frontal region surrounded by significant vasogenic edema. Moderate mass effect was seen over left lateral ventricle with midline shift of approximately 18 - 19 mm towards right side. Patient underwent left fronto-temporo-parietal flap craniectomy with C-patch duraplasty and complete macroscopic tumor removal. Postoperative histopathology report showed features of gliosarcoma, and immunohistochemistry (IHC) showed glial fibrillary acidic protein (GFAP) focal positivity with spindle cells showing vimentin positivity and desmin negativity. Patient was started on adjuvant radiotherapy with concurrent temozolomide followed by six cycles of adjuvant chemotherapy with temozolomide and kept on regular follow-up. After a period of 1.5 years after completion of adjuvant chemotherapy, patient developed recurrence of measuring approximately 2.2 × 1.3 cm in left fronto-temporal region. The lesion appeared as hyperdense enhancing mass on contract-enhanced computed tomography (CECT) scan. The patient underwent re-excision of the recurrent mass and was again started on temozolomide-based chemotherapy with the dose of 250 mg daily for 5 days every 28 days for six cycles and is on follow-up since 9 months.
Case 2
A 35-year-old female presented to neurosurgery department of our institute, with complaints of predominantly left-sided headache, vomiting and right-sided weakness for the last 2 months. There was no associated history of seizures, visual disturbances, fever or loss of consciousness. Magnetic resonance imaging (MRI) of brain showed heterogeneously enhancing mass measuring 4.9 × 8.4 × 4.5 cm with large central necrosis and perilesional edema seen in parieto-occipital lobe (Fig. 1a). A complete metastatic workup was done, which showed no evidence of disease elsewhere. Mass was completely excised in the neurosurgery department (Fig. 1b).
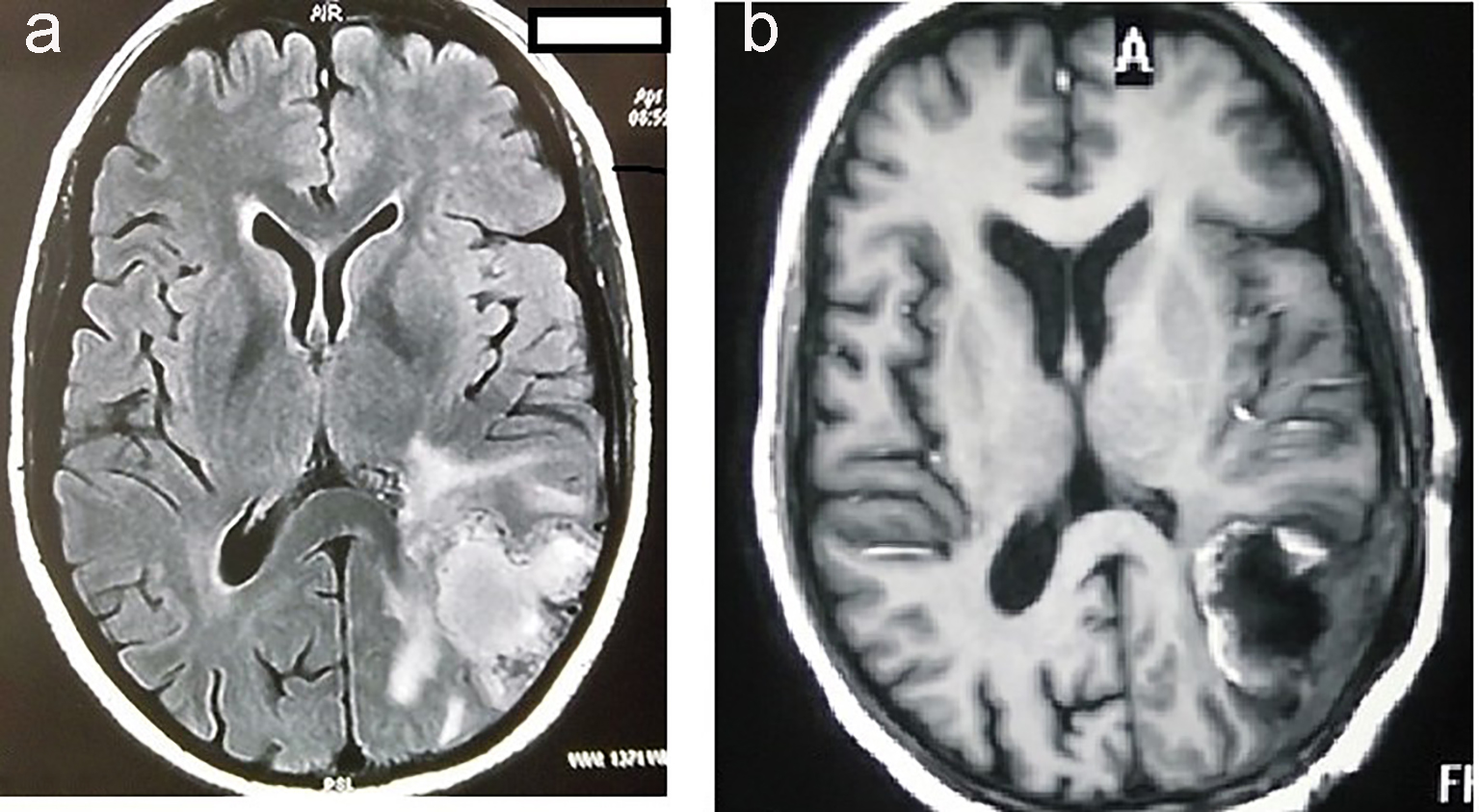 Click for large image | Figure 1. MRI of brain showing hyperintensity in the parieto-occipital lobe of brain (a). MRI of brain showing postoperative defect after complete excision of tumor (b). |
Histopathological examination revealed gliosarcoma (Fig. 2). Patient was referred to our department for postoperative radiotherapy. On examination, patient’s general condition was stable. She was fully conscious, cooperative and well oriented to time and space. Eastern Cooperative Oncology Group (ECOG) performance status was 1. All the baseline blood investigations were normal. Patient was treated by concurrent chemoradiation (60 Gy in 30 fractions) with temozolomide (dose 75 mg/m2) followed by adjuvant chemotherapy with temozolomide (dose 250 mg/m2) for 5 days every 28 days for six cycles. She is now on follow-up since 6 months.
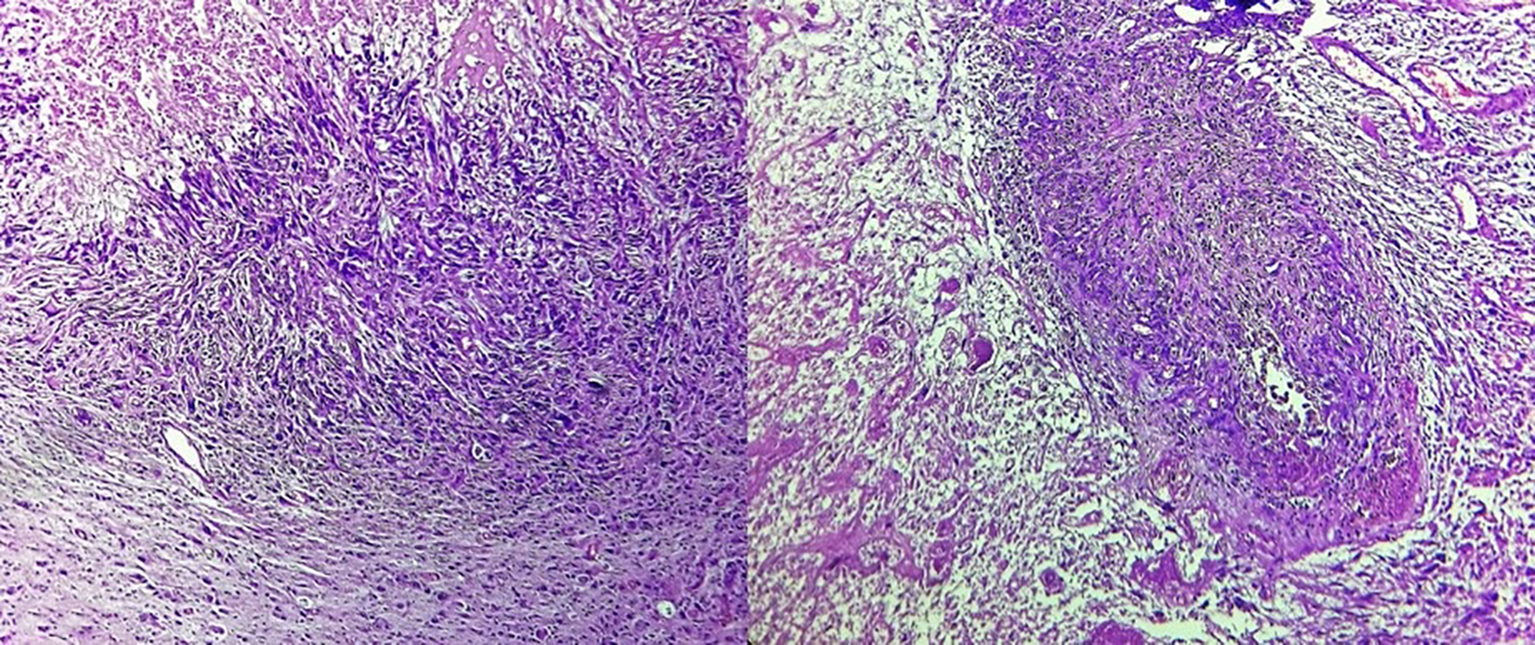 Click for large image | Figure 2. Tumor cells arranged in diffused sheets with areas of necrosis and vascular endothelial proliferation (× 40). Cells show marked nuclear atypia, pleomorphism and hyperchromasia along with atypical mitosis and focal areas of elongated and spindle shaped tumor cells. |
Case 3
A 16-year-old male child was referred to us who had progressed from low grade astrocytoma to gliosarcoma over a long period of 15 years. The child was diagnosed as a case of pilocytic astrocytoma at 6 months of age (in September 2000). The presenting signs and symptoms were progressive increase in head size for 3 weeks with tense and bulging fontanelle, refusal to take feed for 2 days and loss of consciousness for 1 day. CECT scan of head revealed a large cystic space occupying lesion of size 13.7 × 6.7 cm replacing most of the left cerebral hemisphere with an enhancing soft tissue component in its wall suggestive of pilocytic astrocytoma. The infant underwent tapping of tumor cyst followed by left parietal craniotomy and excision of tumor. The postoperative histopathology report showed features suggestive of pilocytic astrocytoma. The child was not planned for any adjuvant radiation and was kept on regular follow-up.
Patient was referred to our center when he developed presenting complaints of headache on and off with multiple episodes of vomiting and disturbance of gait for 1 month at the age of 16 years. On examination, his Glasgow coma score (GCS) was E4M5V6 (15/15) and power was grade 4/5 in right side upper and lower limbs and 5/5 with normal tone and reflexes on the left side. On further investigation, contrast-enhanced magnetic resonance imaging (CEMRI) of brain showed a well-defined extra-axial collection in left fronto-temporo-parieto-occipital region with nodular thickening of the meninges on the medial aspect of collection displaying intense enhancement on post-contrast images. A patchy ill-defined non-enhancing edema was noted in left temporo-parietal region which was further continuous in the posterior fossa extending into left CP angle compressing and displacing the midbrain and pons towards right. The significant mass effect on the left lateral ventricle with midline shift towards right was seen. CECT scan of head was also done which revealed ill-defined multilobulated, cystic solid lesion measuring approximately 10 × 6 × 6.5 cm, with significant enhancement on post-contrast study, involving left occipito-parietal region with soft tissue contents of lesion being hyperdense and cystic contents hypodense. A large hypodense concavo-convex collection was also seen in left fronto-parieto-occipito-temporal region causing significant mass effect in form of displacement of midline structures towards right side by 21 mm causing compression of ventricular system resulting in hydrocephalus without any evidence of hemorrhage. Atrophy of left cerebral hemisphere was seen. Thus, the features on CECT suggested pilocytic astrocytoma with cyst rupture resulting in left subdural collection with obstructive hydrocephalus.
The patient underwent left fronto-temporo-parietal craniotomy with cyst decompression and excision of the tumor. Postoperative histopathology report showed features of high grade malignant tumor with variable morphology in the lateral and medial aspects. On the lateral aspect, the tumor cells showed mainly papillary and perivascular arrangement with cells being round to ovoid in shape with moderate amount of eosinophilic cytoplasm and markedly pleomorphic round nuclei. Brisk mitotic activity and necrosis was also noted. On IHC, the tumor cells showed diffuse positivity with GFAP and were negative for epithelial membrane antigen. On the medial aspect, the tumor cells were predominantly oval to spindle in shape, arranged in interlacing fascicles and bundles, showing marked nuclear atypia and brisk mitoses. Tumor giant cells were interspersed. Occasional papillary configuration was also noted. On IHC, the tumor cells in sarcomatous areas showed diffuse positivity for vimentin, while the glial areas are highlighted by GFAP. CSF cytology showed no presence of malignant cells. Chest X-ray and ultrasound of abdomen were done to assess for any distant metastases, which were reported as normal studies. The child received adjuvant concurrent chemoradiotherapy followed by adjuvant chemotherapy and is being followed up since 4 months.
Case 4
A 23-year-old female patient had developed complaints of giddiness and two episodes of loss of consciousness for 10 days. She also complained of headache and vomiting for 3 days before she was investigated. The patient did not have any neurological deficit. CECT scan showed solid and cystic lesion measuring 4.7 × 5.7 cm in right frontal region with perifocal edema. The solid component of the lesion was contrast enhancing while the cystic part was non-enhancing. Mass effect was seen causing compression over right lateral ventricle with midline shift and subfalcine herniation to left with descending transtentorial herniation. The patient underwent right frontal free bone flap craniotomy and gross total excision of tumor. Histopathology report showed features of mixed spindle cell and glial tumor. The spindle cell component shows a fascicular architecture with moderate nuclear pleomorphism and collagenous stroma. A few atypical mitotic figures were seen. The spindle cell confluent was seen to intimately infiltrate and intermingle with the glial component. The glial component showed mild hypercellularity of astrocytic cells. Oligodendroglial cells were also noted. No necrosis was seen. IHC of tumor cells was immunopositive for vimentin and GFAP. Immunostain for IDH and reticulin was noncontributory. The final impression was gliosarcoma WHO grade 4. The patient was treated with adjuvant radiotherapy with concurrent chemotherapy followed by adjuvant chemotherapy with temozolomide.
| Discussion | ▴Top |
Gliosarcoma was first described by Stroebe in 1895. Gliosarcoma is an uncommon brain tumor and the reported incidence is 1-8% of all malignant gliomas and thus represents an exceptionally rare malignancy [1]. It is a mixed tumor with components of both gliomatous and sarcomatous elements. Since the glial component is histologically similar to typical glioblastoma tissue, it shares similar clinical and genetic features [4]. Exact pathogenesis is unknown, but earlier it has been postulated that the sarcomatous components originated from malignant transformation of hyperplastic blood vessels commonly found in high grade gliomas [5, 6]. Many studies have revealed identical genetic alterations in terms of chromosomal aberrations and cytogenetic imbalances and identical p53 mutations in the gliomatous and sarcomatous components of gliosarcoma, proving their monoclonal origin [7].
Gliosarcoma usually presents in age of 50 - 60 years [2]. However, in this series, all the cases were seen in young age. The median age of presentation was 45 years in a series of 27 cases of primary gliosarcoma [8]. The younger age of distribution was also seen in two other series from India and may be due to geographical variation of presentation of gliosarcoma [9, 10].
Gliosarcomas are generally located in supratentorial region with the commonest site being temporal region followed by the frontal, parietal and occipital lobes [11]. In a series by Kumar et al, 37.5% of patients had primary gliosarcoma located in temporal lobe [10]. Here in one case tumor was located in parieto-occipital lobe which is an uncommon location site for gliosarcoma while in another case, a large cystic mass was seen occupying left fronto-temporo-parietal region with multiple calcific foci in soft tissue components. Though distant metastasis to lung, bone and lymph nodes has been reported in the literature, no systemic metastasis was seen in this series [12].
Treatment for gliosarcoma is similar to GBM [13] as the precise treatment recommendations are still not available due to small number of patients in the available case series and case reports. Thus, tumor resection, postoperative radiation therapy, and chemotherapy with nitrosureas, misonidazole, dacarbazine, mithramycin, ametophterin, thalidomide, temozolomide, irinotecan, vincristine, cisplatin, or doxorubicin are the treatment modalities for gliosarcoma [13]. Treatment with concurrent chemoradiation with temozolomide is suggested to improve overall survival [14]. But the prognosis remains in dismay despite of treatment with the mean survival being 13 months (6.9 - 19.4 months) [3, 15].
Adjuvant radiotherapy dose is generally 60 Gy in 30 fractions over 6 weeks delivered in two phases though hypofractionated radiotherapy (40 Gy/15 fractions) can be given in patients with poor performance status [16]. Phase I planning (46 Gy/23 fractions/4.5 weeks) is done taking into account the initial tumor and edema with sufficient margins followed by boost to tumor bed in phase II planning (14 Gy/7 fractions/1.5 weeks). Adjuvant radiotherapy improves median survival as documented in a study by Perry et al [17]. The adjuvant radiotherapy is added with concurrent chemotherapy temozolomide 75 mg/m2 followed by six cycles of adjuvant chemotherapy with temozolomide 150 - 200 mg/m2 for 5 days every 4 weeks as per Stupp regimen [18]. Stupp regimen results in better survival when compared to RT alone (though not statistically significant) as reported in a study by Salvati et al [7]. In the Indian series of 27 cases of gliosarcoma, the median overall survival (OS) reported was 16.7 months and 2-year OS was 32.6% [8]. Further improvement in survival can be achieved by more conformal dose delivery techniques and finding novel therapies for treatment with the help of molecular studies with more focus on biology of gliosarcoma.
In this series, we have reported four cases of gliosarcoma who have been treated on the lines of GBM. All the cases are being followed up regularly to look into the natural course of the tumor.
Conclusion
Gliosarcoma is a rare central nervous system malignancy. Here we have reported four cases of gliosarcoma. As gliosarcoma is considered as a variant of GBM, it is treated similarly to GBM but optimum treatment recommendations are yet to be defined. Gliosarcoma carries poor prognosis. The studies focusing on cellular and molecular biology of gliosarcoma are required which would aid in identifying new modalities of treatment and bring hope to improve outcomes in patients diagnosed with gliosarcoma.
| References | ▴Top |
- Kozak KR, Mahadevan A, Moody JS. Adult gliosarcoma: epidemiology, natural history, and factors associated with outcome. Neuro Oncol. 2009;11(2):183-191.
doi pubmed - di Norcia V, Piccirilli M, Giangaspero F, Salvati M. Gliosarcomas in the elderly: analysis of 7 cases and clinico-pathological remarks. Tumori. 2008;94(4):493-496.
pubmed - Lutterbach J, Guttenberger R, Pagenstecher A. Gliosarcoma: a clinical study. Radiother Oncol. 2001;61(1):57-64.
doi - Galanis E, Buckner JC, Dinapoli RP, Scheithauer BW, Jenkins RB, Wang CH, O'Fallon JR, et al. Clinical outcome of gliosarcoma compared with glioblastoma multiforme: North Central Cancer Treatment Group results. J Neurosurg. 1998;89(3):425-430.
doi pubmed - Feigin IH, Gross SW. Sarcoma arising in glioblastoma of the brain. Am J Pathol. 1955;31(4):633-653.
pubmed - Khanna M, Siraj F, Chopra P, Bhalla S, Roy S. Gliosarcoma with prominent smooth muscle component (gliomyosarcoma): a report of 10 cases. Indian J Pathol Microbiol. 2011;54(1):51-54.
doi pubmed - Salvati M, Caroli E, Raco A, Giangaspero F, Delfini R, Ferrante L. Gliosarcomas: analysis of 11 cases do two subtypes exist? J Neurooncol. 2005;74(1):59-63.
doi pubmed - Rath GK, Sharma DN, Mallick S, Gandhi AK, Joshi NP, Haresh KP, Gupta S, et al. Clinical outcome of patients with primary gliosarcoma treated with concomitant and adjuvant temozolomide: A single institutional analysis of 27 cases. Indian J Cancer. 2015;52(4):599-603.
doi pubmed - Biswas A, Kumar N, Kumar P, Vasishta RK, Gupta K, Sharma SC, Patel F, et al. Primary gliosarcoma - clinical experience from a regional cancer centre in north India. Br J Neurosurg. 2011;25(6):723-729.
doi pubmed - Kumar P, Singh S, Krishnani N, Datta NR. Gliosarcoma: an audit from a single institution in India of 24 post-irradiated cases over 15 years. J Cancer Res Ther. 2008;4(4):164-168.
doi pubmed - Vukelic Z, Kalanj-Bognar S, Froesch M, Bindila L, Radic B, Allen M, Peter-Katalinic J, et al. Human gliosarcoma-associated ganglioside composition is complex and distinctive as evidenced by high-performance mass spectrometric determination and structural characterization. Glycobiology. 2007;17(5):504-515.
doi pubmed - Weaver D, Vandenberg S, Park TS, Jane JA. Selective peripancreatic sarcoma metastases from primary gliosarcoma. Case report. J Neurosurg. 1984;61(3):599-601.
doi pubmed - Han SJ, Yang I, Tihan T, Prados MD, Parsa AT. Primary gliosarcoma: key clinical and pathologic distinctions from glioblastoma with implications as a unique oncologic entity. J Neurooncol. 2010;96(3):313-320.
doi pubmed - Walker GV, Gilbert MR, Prabhu SS, Brown PD, McAleer MF. Temozolomide use in adult patients with gliosarcoma: an evolving clinical practice. J Neurooncol. 2013;112(1):83-89.
doi pubmed - Han SJ, Yang I, Tihan T, Chang SM, Parsa AT. Secondary gliosarcoma: a review of clinical features and pathological diagnosis. J Neurosurg. 2010;112(1):26-32.
doi pubmed - Kumar N, Bhattacharyya T, Chanchalani K, Shalunke P, Radotra BD, Yadav BS. Impact of changing trends of treatment on outcome of cerebral gliosarcoma: A tertiary care centre experience. South Asian J Cancer. 2015;4(1):15-17.
doi pubmed - Perry JR, Ang LC, Bilbao JM, Muller PJ. Clinicopathologic features of primary and postirradiation cerebral gliosarcoma. Cancer. 1995;75(12):2910-2918.
doi - Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.