

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website http://www.wjon.org |
Case Report
Volume 10, Number 6, December 2019, pages 226-230
Bone Marrow Features in Patients With Acute Myeloid Leukemia Treated With Novel Targeted Isocitrate Dehydrogenase 1/2 Inhibitors
Ashley Hagiyaa, Poorva Vaidyab, Tarek Khedroc, Bassam Yaghmourd, Imran Siddiqia, George Yaghmourc, e
aDepartment of Pathology, Keck School of Medicine of University of Southern California, Los Angeles, CA 90033, USA
bInternal Medicine, Keck School of Medicine of University of Southern California, Los Angeles, CA 90033, USA
cJane Anne Nohl Division of Hematology and Center for the Study of Blood Diseases, Keck School of Medicine of University of Southern California, Los Angeles, CA 90033, USA
dPulmonary Critical Care, Keck School of Medicine of University of Southern California, Los Angeles, CA 90033, USA
eCorresponding Author: George Yaghmour, Jane Anne Nohl Division of Hematology and Center for the Study of Blood Diseases, Keck School of Medicine of University of Southern California, 1441 Eastlake Ave, Los Angeles, CA 90033, USA
Manuscript submitted September 5, 2019, accepted September 23, 2019
Short title: Pathologic Features in AML and IDH Inhibitors
doi: https://doi.org/10.14740/wjon1231
| Abstract | ▴Top |
This case report aimed to review the bone marrow features of patients with acute myeloid leukemia (AML) treated with isocitrate dehydrogenase 1/2 (IDH1/2) inhibitors. Five patients with AML treated with an IDH1/2 inhibitor were identified and retrospectively reviewed. We described the morphologic and immunophenotypic findings in the bone marrow, as well as ancillary study results. Two patients showed a hypercellular bone marrow with morphologic and immunophenotypic differentiation of blasts. The bone marrow of one patient displayed a hypoplastic phase. Four of the five patients demonstrated unusual morphologic and/or immunophenotypic populations, including basophilia with mild alterations on the myeloid blasts, a small subset of blasts with expression of T-cell markers not seen in the original immunophenotype, a cluster of differentiation 117 (CD117)-positive progenitor population with erythroid differentiation, and another population reminiscent of erythroid differentiation. Unusual morphologic and immunophenotypic populations can be seen in the bone marrows of patients treated with IDH1/2 inhibitors in the presence or absence of definite residual disease. The significance of these populations is uncertain, but further studies could be helpful to understand the meaning of these findings.
Keywords: Acute myeloid leukemia; Isocitrate dehydrogenase inhibitors; Bone marrow microenvironment
| Introduction | ▴Top |
Isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase 2 (IDH2) encode cytoplasmic/peroxisomal IDH1 and mitochondrial IDH2, respectively, which catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) [1]. IDH1 and IDH2 mutations are seen in approximately 6-16% and 8-19% of acute myeloid leukemia (AML) patients, respectively [2, 3]. Mutations involve a single amino acid substitution at an arginine (R) residue, most commonly R132 in IDH1 and R140 or R172 in IDH2, resulting in neomorphic enzyme activity and accumulation of R-2-hydroxyglutarate (R-2-HG) [1, 4, 5]. R-2-HG competitively inhibits α-KG dependent enzymes, leading to DNA hypermethylation, increased repressive histone methylation, and impaired hematopoietic differentiation [4, 5].
Ivosidenib and enasidenib are oral, small molecule inhibitors of mutant-IDH1 and mutant-IDH2 enzymes, respectively [6]. These inhibitors reduce 2-HG to normal levels and promote differentiation of leukemic hematopoietic progenitor cells without an intervening period of bone marrow aplasia or hypoplasia [5, 6]. This leads to maturation of the blasts to functional neutrophils, which retain the IDH1/2 mutation [5]. Ivosidenib and enasidenib are effective salvage therapies and may serve as a bridge to transplant. They are also noncytotoxic, making them more tolerable for older patients [7].
Bone marrow evaluation is essential in the initial workup and monitoring of patients with AML. With the advent of new noncytotoxic therapies, it is important to be aware of their effects on the bone marrow, especially when assessing for residual disease. Here we present the bone marrow findings from five patients with AML, before and after treatment with ivosidenib or enasidenib.
| Case Reports | ▴Top |
After approval from the Institutional Review Board (IRB), five patients with AML treated with an IDH1/2 inhibitor were identified and retrospectively reviewed. We described the bone marrow morphologic and immunophenotypic findings, as well as the results of ancillary studies. A chart review of the electronic medical record was also performed.
In total, five relapsed/refractory AML patients treated with IDH1/2 inhibitors were identified. A summary of the findings is described below and in Supplementary Material 1 (www.wjon.org).
Case 1
The patient was a 56-year-old woman with no past medical history. She was diagnosed with AML with monocytic differentiation in December 2017. At that time the bone marrow was normocellular with 60% blasts. Cytogenetics showed trisomy 8 in 17 of 20 cells. Molecular studies demonstrated mutations in the IDH2, DNMT3A and BCOR genes (variant allele fractions (VAFs) not available). The patient’s disease proved to be refractory to 7 + 3, as well as high-dose cytarabine (HiDAC). She was started on enasidenib and then referred to our institution for allogeneic stem cell transplant evaluation.
A bone marrow biopsy was performed approximately 3 months after the initiation of enasidenib (Supplementary Material 1, www.wjon.org). The marrow was hypo- to normocellular with basophilia and monocytosis, but no definite increase in blasts. Minimal residual disease (MRD) flow cytometry performed at the University of Washington showed proportionally increased basophils (4.4% of leukocytes) with mild immunophenotypic alterations identified on myeloid blasts (3.8% of leukocytes) including mildly increased cluster of differentiation 4 (CD4), CD33, and CD123, and mildly decreased expression of human leukocyte antigen-DR (HLA-DR) isotype, and CD7 expression on a minor subset; monocytes were also proportionally increased (17.3%) with myeloid and monocytic left-shift. The karyotype was normal and fluorescence in situ hybridization (FISH) did not detect trisomy 8. However, given these mild alterations, a repeat biopsy was performed 1 month later. The bone marrow was hypocellular with granulocytic left-shift and monocytosis. Definite blasts comprised less than 5% of total cells. Flow cytometry again showed proportionally increased basophils with similar immunophenotypic alterations identified on the blasts as previously described. IDH1/2 mutation analysis was negative (assay sensitivity 10-15%).
She underwent transplant in August 2018, approximately 5 months after starting enasidenib. A day-100 bone marrow biopsy showed a hypo- to normocellular bone marrow with less than 5% blasts. No abnormal myeloid blast population was identified by MRD flow cytometry. She continued to be in complete remission (CR), transfusion-independent, without significant graft-versus-host disease.
Case 2
The patient was a 74-year-old man who presented to his primary care physician after feeling unwell for some time, and was found to be pancytopenic. A bone marrow biopsy was subsequently performed. The marrow was normocellular with about 20% blasts, dysgranulopoiesis, and dysmegakaryocytopoiesis, consistent with AML with myelodysplasia-related changes (AML-MRCs). The blasts expressed CD13 dim, CD33 dim, CD34, CD36 partial, CD38, CD117, HLA-DR, and myeloperoxidase (MPO) partial by flow cytometry. Chromosome analysis demonstrated a normal male karyotype. Molecular studies revealed mutations in the IDH2 R140 and cohesion subunit SA-2 (STAG2) genes with a VAF of 42% and 84%, respectively. He was treated with liposomal cytarabine-daunorubicin. Day-14 and day-21 bone marrow evaluations revealed residual disease with 15-20% blasts. He was pancytopenic. Upon count recovery, a subsequent bone marrow biopsy, on day 54, showed 2.7% residual blasts.
Subsequently, enasidenib was started, and a bone marrow biopsy performed about 1 month later was mildly hypocellular (30%) with 6.1% blasts showing a similar immunophenotype to that described at diagnosis, by flow cytometry performed at the University of Washington. In addition, a small subset (0.3%) of blasts were CD34-positive with expression of cCD3, CD5 dim, CD7 bright, and negative for CD4, CD14, CD16, CD19, CD56, and CD64. Enasidenib was briefly discontinued for 1 month. Subsequent bone marrow evaluations, 1 and 5 months after restarting enasidenib, again demonstrated a small subset of CD34-positive blasts with bright CD7 and intermediate CD5 expression along with a minor population of residual blasts with the original immunophenotype. The patient continued to be in hematologic CR, and transfusion-independent, with normal blood counts. He has been following up with his local hematologist without report of relapse.
Case 3
The patient was a 66-year-old woman with a past medical history of diabetes, rheumatoid arthritis, hepatitis B, and treated hepatitis C. She initially presented to an outside hospital with excessive fatigue, rash, and arthralgias, which were attributed to rheumatoid arthritis. However, the fatigue worsened, and she developed easy bruising. At that time, she returned to the emergency department where she was found to be pancytopenic and have circulating blasts. She was transferred to our institution for a higher level of care.
The initial bone marrow biopsy was hypercellular with 32% blasts. The blasts were large with irregular to clefted nuclei, fine chromatin, variably prominent nucleoli, and scant amounts of basophilic cytoplasm with punched out cytoplasmic vacuoles. By flow cytometry, the blasts expressed CD13 dim, CD34, CD117, HLA-DR, and MPO dim. Cytogenetics demonstrated a complex karyotype. Molecular studies revealed pathogenic mutations in the IDH2 R140 and tumor protein 53 (TP53) genes with a VAF of 45% and 57%, respectively. Genomic alterations of uncertain significance were also detected in the DNMT3A and BCOR genes with a VAF of 46% and 51%, respectively. The findings were consistent with AML-MRC. She was treated with a 10-day course of decitabine. After three cycles of decitabine, a subsequent bone marrow was normocellular with 10-15% blasts, consistent with residual AML.
Enasidenib was started, and a bone marrow biopsy was performed approximately 1 month later. The marrow was hypercellular with less than 5% blasts. Flow cytometry did not identify a definite abnormal myeloid blast population, but there was a CD117+ progenitor population with erythroid differentiation. However, the karyotype was complex, indicating persistent disease. Approximately 2 months later, the patient relapsed with 90% involvement of a hypercellular bone marrow. IDH2 R140, TP53, DNMT3A, and BCOR mutations were still detected and the karyotype remained complex. One month later, the bone marrow continued to be hypercellular with morphologic evidence of partial blast differentiation to an immature myelomonocytic form showing punched out vacuoles similar to those seen in the original blasts (Fig. 1). In addition, the blasts demonstrated immunophenotypic evidence of differentiation with decreased to absent CD34 and CD117 expression by flow cytometry and immunohistochemistry. IDH2 R140 and TP53 mutations persisted. Unfortunately, the patient expired shortly after.
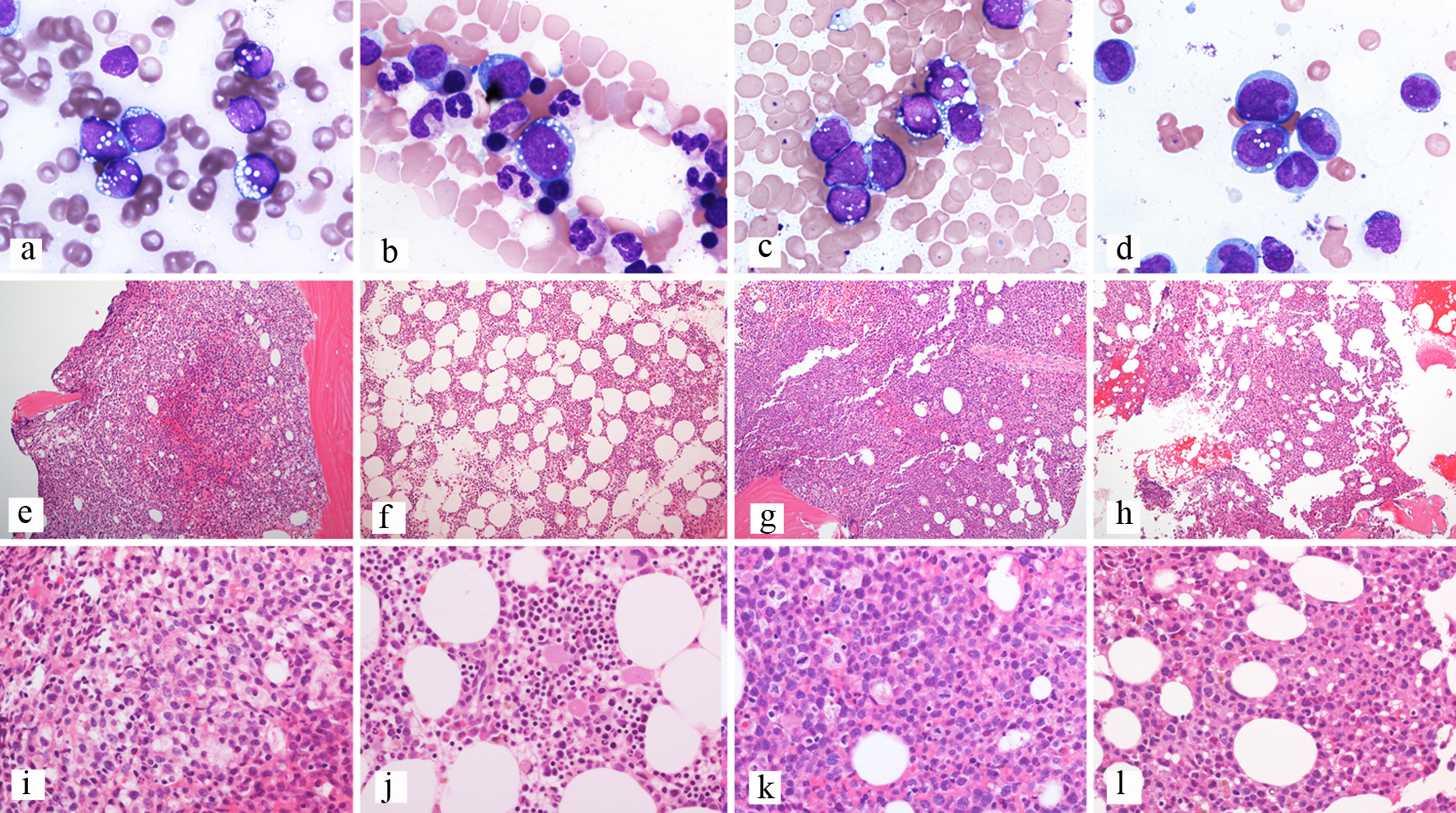 Click for large image | Figure 1. Composite of bone marrow biopsies from patient 3. (a, e, i) Bone marrow biopsy at diagnosis; (a) Aspirate showing blasts with cytoplasmic vacuoles, Wright-Giemsa stain, 100 × objective; (e) Trephine biopsy showing a hypercellular bone marrow, H&E-stained section, 10 × objective; (i) Trephine biopsy showing sheets of blasts, H&E-stained section, 40 × objective. (b, f, j) Bone marrow biopsy after enasidenib; (b) Aspirate showing rare erythroid precursors with cytoplasmic vacuoles and no definite blasts, Wright-Giemsa stain, 100 × objective; (f) Trephine biopsy showing a hypercellular bone marrow, H&E-stained section, 10 × objective; (j) Trephine biopsy showing trilineage hematopoiesis with erythroid hyperplasia, H&E-stained section, 40 × objective. (c, g, k) Bone marrow biopsy at relapse; (c) Aspirate showing blasts with cytoplasmic vacuoles, Wright-Giemsa stain, 100 × objective; (g) Trephine biopsy showing a hypercellular bone marrow, H&E-stained section, 10 × objective; (k) Trephine biopsy showing sheets of blasts, H&E-stained section, 40 × objective. (d, h, l) Bone marrow biopsy after enasidenib; (d) Aspirate showing blasts with partial differentiation and similar cytoplasmic vacuoles, Wright-Giemsa stain, 100 × objective; (h) Trephine biopsy showing a hypercellular bone marrow, H&E-stained section, 10 × objective; (l) Trephine biopsy showing sheets of immature cells, H&E-stained section, 40 × objective. H&E: hematoxylin and eosin. |
Case 4
The patient was a 63-year-old man with no past medical history who presented to an outside hospital with left-sided abdominal pain and nausea. He was found to have an elevated white count and anemia.
A bone marrow biopsy was performed and he was diagnosed with AML, not otherwise specified. Chromosome analysis showed trisomy 8. Molecular studies demonstrated pathogenic alterations in the IDH2 R140, splicing factor arginine/serine-rich 2 (SRSF2), and STAG2 genes with a VAF of 44%, 44%, and 60%, respectively. An alteration of uncertain significance was also detected in the translocation-Ets-leukemia virus (ETV6) gene with a VAF of 46%.
He was treated with 7 + 3. Day-14 bone marrow biopsy showed persistent disease with marrow cellularity of 20% and 30-40% blasts. So, salvage chemotherapy using 5 + 2 was given. Upon count recovery, day-48 bone marrow biopsy showed CR. On day 70, consolidation therapy was started using 1,500 mg/m2 intermediate dose cytarabine (IDAC) every 12 h on days 1, 3, and 5. This course of consolidation was complicated by septic shock with multiorgan failure from a pulmonary Mycobacterium avium complex (MAC) infection that was treated with ethambutol, rifabutin, and azithromycin. The patient recovered. Given the patient’s critical status and the organ dysfunction after the first course of consecutive therapy, and given the HOVON97 randomized Phase III data showing improvement in disease-free-survival for older AML patients in CR after at least two cycles of intensive chemotherapy [8], the patient was placed on a hypomethylating agent (decitabine) starting on day 125. Since the patient had an IDH2 mutation and could not continue the standard-of-care consolidation, his treating physician decided to add enasidenib to the decitabine 3 months later. Three months after starting this combined therapy, MRD flow cytometry detected 0.2% residual blasts involving a hypocellular bone marrow. Two months later, a day-340 bone marrow biopsy showed blasts comprising about 5% of cells, and he was started on a combination of gemtuzumab with enasidenib. He relapsed 2 months later, and the patient was subsequently enrolled in a clinical trial. At that time enasidenib was discontinued.
Case 5
The patient was a 60-year-old woman with a past medical history of hypothyroidism. She presented to an outside hospital with easy bruising and gingival bleeding. A complete blood count (CBC) demonstrated leukocytosis, anemia, and thrombocytopenia. The initial bone marrow biopsy showed 92% blasts (Supplementary Material 1, www.wjon.org). There were too few nonblast elements to assess for morphologic dysplasia. The karyotype was complex. Molecular studies detected mutations in the TP53, DNMT3A and IDH1 R132 genes with a VAF of 94%, 47% and 48% (two mutations), and 49%, respectively. The overall findings were consistent with AML-MRC.
The patient started induction therapy with liposomal cytarabine-daunorubicin and on day 14 showed persistent disease of 80% cellularity with 98% blasts. She was enrolled on a clinical trial, but she continued to progress and was found to have 40% peripheral blood blasts after two cycles. The flow cytometry for this peripheral blood blasts confirmed leukemia progression. Thus, the patient was started on a hypomethylating agent, decitabine, for two cycles. After the second cycle, the peripheral blasts measured 80% and flow cytometry confirmed progression. Thus, venetoclax was added to decitabine [9]. However, venetoclax was poorly tolerated by the patient, resulting in significant gastrointestinal (GI) symptoms, and was discontinued. Decitabine was continued as a single agent for one more cycle, yet the patient continued to progress, measured by the peripheral blasts. Given the IDH1 mutation, she was switched to ivosidenib. Shortly after, she developed increasing oxygen requirements and a chest X-ray showed bilateral infiltrates. The findings were concerning for differentiation syndrome. Ivosidenib was discontinued for a few days and dexamethasone was given per the label. Once the patient recovered, ivosidenib was resumed at a lower dose, and titrated up.
A bone marrow biopsy was performed approximately 4 weeks after initiation of IDH1 inhibitor therapy and was hypercellular with less than 5% blasts. MRD flow cytometry detected an unusual population of myeloid blasts representing 0.8% of total white cells and showing discrete expression of increased CD38, CD71, high-level CD117, and HLA-DR without CD13, CD15, CD33, CD64, or CD123. The immunophenotype was reminiscent of early erythroid precursors but unusual for the high level of HLA-DR. Cytogenetics showed a persistent complex karyotype.
On day 164, the patient was admitted again for differentiation syndrome, and ivosidenib was held due to hemoptysis. Patient recovered and the ivosidenib was restarted. A day-171 bone marrow biopsy (3 months from ivosidenib initiation) showed 10% cellularity and < 5% blasts. MRD flow cytometry detected no abnormal blast population and found a normal female cytogenetic karyotype. She underwent haploidentical stem cell bone marrow transplantation in January 2019. The patient is currently 8 months post-transplant and is in CR with no graft-versus-host disease.
| Discussion | ▴Top |
IDH1/2 inhibitors are noncytotoxic therapies for AML which cause differentiation of leukemic hematopoietic progenitors. Literature describing the morphologic and immunophenotypic findings seen in the bone marrow of patients treated with these therapies is limited.
The first-in-human phase 1/2 study assessing the properties of enasidenib described a decrease in blasts and the appearance of mature myeloid forms with a normal immunophenotype and no intervening marrow aplasia or hypoplasia [5]. Mature granulocytes from patients in remission retained the IDH2 mutation and cytogenetic abnormalities [5]. An ivosidenib study also showed that the drug induces myeloid differentiation and trilineage hematopoietic recovery without an intervening period of bone marrow aplasia [6]. In our series, patient 3 displayed morphologic differentiation of blasts to immature myelomonocytic cells, as well as immunophenotypic differentiation to a more mature, but still abnormal phenotype. There was also persistence of the IDH2 mutation and complex karyotype. These findings were consistent with the study conclusions. Unlike patients 3 and 5, which showed hypercellular bone marrows without intervening aplasia or hypoplasia, patient 1 did demonstrate hypoplasia of the marrow while being treated with an IDH inhibitor.
One center described their experience with IDH2 inhibitors [10]. In their abstract, three of four patients showed a markedly hypercellular bone marrow with sheets or large aggregates of myeloblasts. Two of the cases displayed abnormal morphology at the end of additional cycles of therapy. One case showed a markedly hypercellular bone marrow with myeloid expansion and increased reticulin fibrosis reminiscent of a myeloproliferative disorder. Another case was also markedly hypercellular with marked myeloid and megakaryocytic hyperplasia and trilineage dysplasia. Although patients 3 and 5 demonstrated hypercellular bone marrows after IDH1/2 inhibitor therapy, other features of a myeloproliferative disorder were not observed in our series.
In our series, one patient (patient 1) showed unusual morphologic findings, including increased basophils without definite blasts, and bone marrow hypoplasia. Additionally, four of five patients displayed atypical immunophenotypic findings, including proportionally increased basophils with mild alterations on myeloid blasts (patient 1), a small subset of blasts with expression of T-cell markers not seen in the original immunophenotype (patient 2), a progenitor population with erythroid differentiation (patient 3), and a population of myeloid blasts reminiscent of erythroid precursors but with unusually high HLA-DR (patient 5). Given the heterogeneity of leukemic blasts [11], it may not be that surprising that MRD flow cytometry detected minor phenotypic alterations, as our MRD flow cytometry was sent to a reference laboratory where they incorporated the different from normal approach with leukemia associated immunophenotype (LAIP). This approach is less subjective to the effects of blast heterogeneity, phenotypic drift, and clonal selection or evolution [11]. Whether or not the differentiation effect of IDH1/2 inhibitors specifically gives rise to these unusual populations cannot be said with certainty.
Interestingly, patients 2, 3, and 5 were all diagnosed with AML-MRC, and two showed a complex karyotype. This may suggest the possibility that the underlying biologics of a myelodysplasia-related AML could be more prone to these alterations.
Clinicians should be aware that the normal effects of IDH inhibitors on the bone marrow generally do not elicit an intervening marrow aplasia or hypoplasia and induce blast maturation. These mature myeloid forms should display a normal immunophenotype. The significance of phenotypically unusual populations, as observed in this series, is unknown. However, these patients should be followed closely because relapses can occur shortly after identification of these subsets.
Moreover, bone marrow assessment for residual disease can be difficult, especially when the effects of novel therapies are not well-described and not well-recognized. We described unusual morphologic and immunophenotypic findings seen in the bone marrows of patients treated with IDH1/2 therapy. The overall significance of these findings is uncertain. Our case series is limited in number, and further studies could be helpful in ascertaining the meaning of these populations to better predict subsequent outcomes.
| Supplementary Material | ▴Top |
Supplementary Material 1. Bone marrow features.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
The authors do not have any conflict of interest.
Informed Consent
Not applicable.
Author Contributions
AH, PV, TK, BY, IS and GY performed the research, data analysis, and wrote the manuscript. GY supervised the study.
| References | ▴Top |
- Rakheja D, Konoplev S, Medeiros LJ, Chen W. IDH mutations in acute myeloid leukemia. Hum Pathol. 2012;43(10):1541-1551.
doi pubmed - Ragon BK, DiNardo CD. Targeting IDH1 and IDH2 Mutations in Acute Myeloid Leukemia. Curr Hematol Malig Rep. 2017;12(6):537-546.
doi pubmed - Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders JE, Zeilemaker A, van Putten WJ, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010;116(12):2122-2126.
doi pubmed - Amatangelo MD, Quek L, Shih A, Stein EM, Roshal M, David MD, Marteyn B, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017;130(6):732-741.
doi pubmed - Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, Stone RM, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722-731.
doi pubmed - DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, Swords R, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386-2398.
doi pubmed - Stein EM, DiNardo CD, Fathi AT, Pollyea DA, Stone RM, Altman JK, Roboz GJ, et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood. 2019;133(7):676-687.
doi pubmed - Huls G, Chitu D, Havelange V, Jongen-Lavrencic M, van de Loosdrecht A, Biemond B, et al. Randomized maintenance therapy with azacitidine (vidaza) in older patients (≥ 60 years of age) with acute myeloid leukemia (AML) and refractory anemia with excess of blasts (RAEB, RAEB-t). Results of the HOVON97 phase III randomized multicentre study (EudraCT 2008-001290-15). Blood. 2017;130(Suppl 1);463.
- Aldoss I, Yang D, Aribi A, Ali H, Sandhu K, Al Malki MM, Mei M, et al. Efficacy of the combination of venetoclax and hypomethylating agents in relapsed/refractory acute myeloid leukemia. Haematologica. 2018;103(9):e404-e407.
doi pubmed - Yang J, Gabali AM. Morphologic assessment of bone marrow samples in AML patients treated with inhibitors to IDH1 and IDH2 - a single center experience [abstract]. Blood. 2017;130(suppl):5068.
- Ouyang J, Goswami M, Tang G, Peng J, Ravandi F, Daver N, Routbort M, et al. The clinical significance of negative flow cytometry immunophenotypic results in a morphologically scored positive bone marrow in patients following treatment for acute myeloid leukemia. Am J Hematol. 2015;90(6):504-510.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.