

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Case Report
Volume 13, Number 1, February 2022, pages 38-47

Non-GCB Diffuse Large B-Cell Lymphoma With an Atypical Disease Course: A Case Report and Clinical Exome Analysis
Svetlana Tsygankovaa, d, e, Daria Komovaa, d, Eugenia Boulyginaa, Natalia Slobodovaa, Fedor Sharkoa, Sergey Rastorgueva, Maria Gladysheva-Azgaria, Daria Korolevab, Anna Smol’yaninovab, Svetlana Tatarnikovab, Tatiana Obuchovab, Artem Nedoluzhkoc, Nelli Gabeevab, Eugene Zvonkovb
aNational Research Center “Kurchatov Institute”, 123182 Moscow, Russia
bNational Medical Hematology Research Center, 125167 Moscow, Russia
cMoscow Healthcare Department, Mental-Health Clinic No. 1 Named After N.A. Alexeev, 115191 Moscow, Russia
dThese authors contributed equally.
eCorresponding Author: Svetlana Tsygankova, National Research Center “Kurchatov Institute”, 123182 Moscow, Russia
Manuscript submitted November 17, 2021, accepted December 31, 2021, published online February 8, 2022
Short title: A Case of Non-GCB DLBCL
doi: https://doi.org/10.14740/wjon1436
| Abstract | ▴Top |
Diffuse large B-cell lymphoma (DLBCL) is the most common lymphoid tumor among other non-Hodgkin lymphomas (30-40% of all cases). This type of lymphoma is characterized by significant differences in treatment response and the heterogeneity of clinical traits. Approximately 60% of patients are cured using standard chemotherapy (CT), while in 10-15% of cases, the tumor is characterized by an extremely aggressive course and resistance to even the most high-dose programs with autologous stem cell transplantation (auto-SCT). The activated B-cell (ABC) subtype of DLBCL is characterized by poor prognosis. Here, we describe a clinical case of diffuse ABC-DLBCL with an atypical disease course. Complete remission was achieved after four courses of CT, followed by autologous hematopoietic stem cell transplantation (auto-HSCT). However, early relapse occurred 2 months after the completion of treatment. According to the results of cytogenetic studies, significant chromosome breakdowns were observed. Exome sequencing allowed for the detection of several novel mutations that affect components of the NOTCH2 and NF-κB signaling pathways, a number of epigenetic regulators (KMT2D, CREBBP, EP300, ARID1A, MEF2B), as well as members of the immunoglobulin superfamily (CD58 and CD70). Whether these mutations were the result of therapy or were originally present in the lymphoid tumor remains unclear. Nevertheless, the introduction of genomic technologies into clinical practice is important for making a diagnosis and developing a DLBCL treatment regimen with the use of targeted drugs.
Keywords: Lymphoma; Diffuse large B-cell lymphoma; ABC type; Exome; Sequencing; NOTCH2 pathway; NF-κB pathway; R-mNHL-BFM-90 protocol
| Introduction | ▴Top |
Diffuse large B-cell lymphoma (DLBCL) is a lymphoid tumor that represents one of the most complex therapeutic problems in hematology due to the heterogeneity in both clinical characteristics and the response to therapy. The combination of cyclophosphamide, hydroxydaunorubicin, hydrochloride, vincristine, prednisone (CHOP) with rituximab (R-CHOP), the first anti-cluster of differentiation (CD)20 monoclonal antibody, has changed the outcome of DLBCL, becoming the new standard of care. While about 60% of patients are cured by standard chemotherapy (CT), in 10-15% of cases, the tumor is characterized by an extremely aggressive course and resistance to even the most high-dose programs involving autologous hematopoietic stem cell transplantation (auto-HSCT) [1-4].
Recent studies of gene expression, mutational status, and chromosomal abnormalities have clearly demonstrated the biological diversity of DLBCL and provide a basis for revising existing classifications and developing new differentiated treatment protocols. For example, the wider implementation of fluorescence in situ hybridization (FISH) led to double-hit lymphoma (DHL) being singled out from the general group of DLBCL as a separate entity, and it is characterized by the combined rearrangement of the MYC, BCL2, and/or BCL6 genes [5]. There are currently no effective CT programs for this group of patients. Gene expression profiling of activated B-cell (ABC)-DLBCL cells in the postgerminal stage of differentiation, conducted by Alizadeh et al, allowed for a prognostically unfavorable molecular variant of DLBCL to be identified [6]. This subtype is characterized by constitutional activation of the B-cell receptor signaling pathway and the transcription factor NF-κB (nuclear factor kappa-light chain-enhancer of activated B cells). According to the International Prognostic Index (IPI), the 5-year non-progressive survival (NPS) of ABC-DLBCL patients in the high-risk group was only 28% following standard CT [2, 7]. The high expression of multiple myeloma 1 (MUM1) protein, characteristic of this DLBCL subtype, became the basis for the introduction of lenalidomide into the treatment regimen [8-11]. It is also known that lenalidomide as an immunomodulatory drug, increases the T-cell immune response against B-cell tumors [12].
Until recently, most molecular genetic studies focused on the study of single gene abnormalities, such as in c-MYC, MYD88, EZH2, CREBBP, FOXP1, and TP53 [13-18]. Mutations of some of these genes are a powerful negative prognostic factor, regardless of other clinical and laboratory factors.
However, aberration variants and combinations of different genes make it difficult to accurately assess their clinical significance and identify the group of patients with the highest risk of developing resistance/relapse. With the introduction of next-generation sequencing (NGS) technology, it became possible to comprehensively analyze a much larger amount of data and create a “molecular portrait” of the tumor. As a result of whole exome sequencing (WES) of tumor samples from 304 patients with DLBCL, five molecular clusters were identified, each of which was determined by considering the spectrum of the most frequent genetic breakdowns, commonly used signaling pathways, and prognosis [19, 20]. Of these, clusters C2 (cases with biallelic inactivation of the TP53 gene) and C5 (cases of ABC-DLBCL in combination with 18q/BCL2 amplification and mutations of the CD79B, MYD88, PIM1, ETV6 genes) were associated with the worst prognosis.
However, it is not always possible to accurately determine the particular molecular subtype of a tumor. Tumor cells can simultaneously engage several signaling pathways and acquire additional mutations during their evolution. Therefore, research clarifying the molecular subtypes and their number is ongoing. Given that most of these studies are conducted on groups of patients who received standard CT, the results vary significantly [21]. It seems rational to determine the most significant driver mutations in tumors of DLBCL patients from the high-risk group, which are resistant to the most intensive treatment programs.
In our clinic, the use of the intensive block program R-mNHL-BFM-90 resulted in the improved treatment of a group of DLBCL patients with unfavorable prognosis factors [22]. A retrospective evaluation of the results showed that in most cases of treatment failure, the tumor belonged to the postgerminal immunohistochemical variant of DLBCL (non-germinal center B-cell (non-GCB)-DLBCL), a molecular analog of the ABC-DLBCL subtype [23].
In this article, we present a clinical case description and full-exome sequencing analysis of tumor material from a high-risk patient with non-GCB-DLBCL, who relapsed after first remission following intensive therapy in combination with lenalidomide and high-dose consolidation.
| Case Report | ▴Top |
The male patient, 55 years old, presented with enlargement of the cervical nodes, left palatine tonsil, difficulty swallowing, night sweats, and fever. Positron emission tomography combined with computed tomography (PET/CT) examination revealed an increase in the size of the left palatine tonsil, cervical, intra-abdominal, inguinal, and femoral lymph nodes up to 5 cm in diameter, and a paratracheal conglomerate of lymph nodes up to 9 cm (SUVmax up to 21.2). The activity of lactate dehydrogenase (LDH) was 557 U/L. No data were obtained for bone marrow damage. Immunomorphological examination of the lymph node biopsy revealed diffuse proliferation of medium- and large-sized cells with round-oval, multi-lobed nuclei each containing 2 - 3 nucleoli. The tumor cells expressed CD20, BCL-2, and MUM1 and were negative for CD10, BCL, and Ki67 in 80-90% of the population. Standard cytogenetic examination (SCE) of lymph node cells did not reveal chromosomal aberrations. Translocation involving the BCL6/3q27 gene locus and an additional signal from the BCL2/18q21 gene locus were observed in 30% of the nuclei using FISH. The deletion of 17p13 and monosomy of chromosome 17 were not detected. Thus, the diagnosis was established as non-GCB type DLBCL, BCL-2 positive.
Four courses of the R-mNHL-BFM-90 program with lenalidomide were conducted. After three courses, 19 × 106/kg CD34+ cells were harvested. When staging after the fourth course, residual tumor tissue with low metabolic activity (SUVmax 2.4, DS3) remained in the jugular lymph node, according to PET/CT data. For the purpose of consolidation, auto-HSCT was performed.
When the patient was examined 2 months after the end of therapy, he had subfebrile temperature, increased LDH activity (to 670 U/L), and lymph nodes that were enlarged on the right. The metabolically active cervical, iliac, inguinal and femoral lymph nodes, and left tonsil (SUVmax 30.74) were characterized according to the results of PET/CT (Fig. 1). Disease relapse was confirmed based on histological examination of the cervical lymph node sample. Karyotype analysis revealed a clone with multiple subclones, complex karyotype disorders (> 20) including derivatives of chromosomes 3, 14, and 17 (with translocations of BCL6, IGH gene loci, and deletion of TP53), and dicentric marker chromosomes (Fig. 2). Using FISH, translocations involving loci in the BCL6/3q27 and IGH/14q32 genes were detected, along with two additional signals from loci in the IGH/14q32 gene, an additional signal from a locus in the BCL2/18q21 gene, and a TP53/17p13 deletion (Fig. 3).
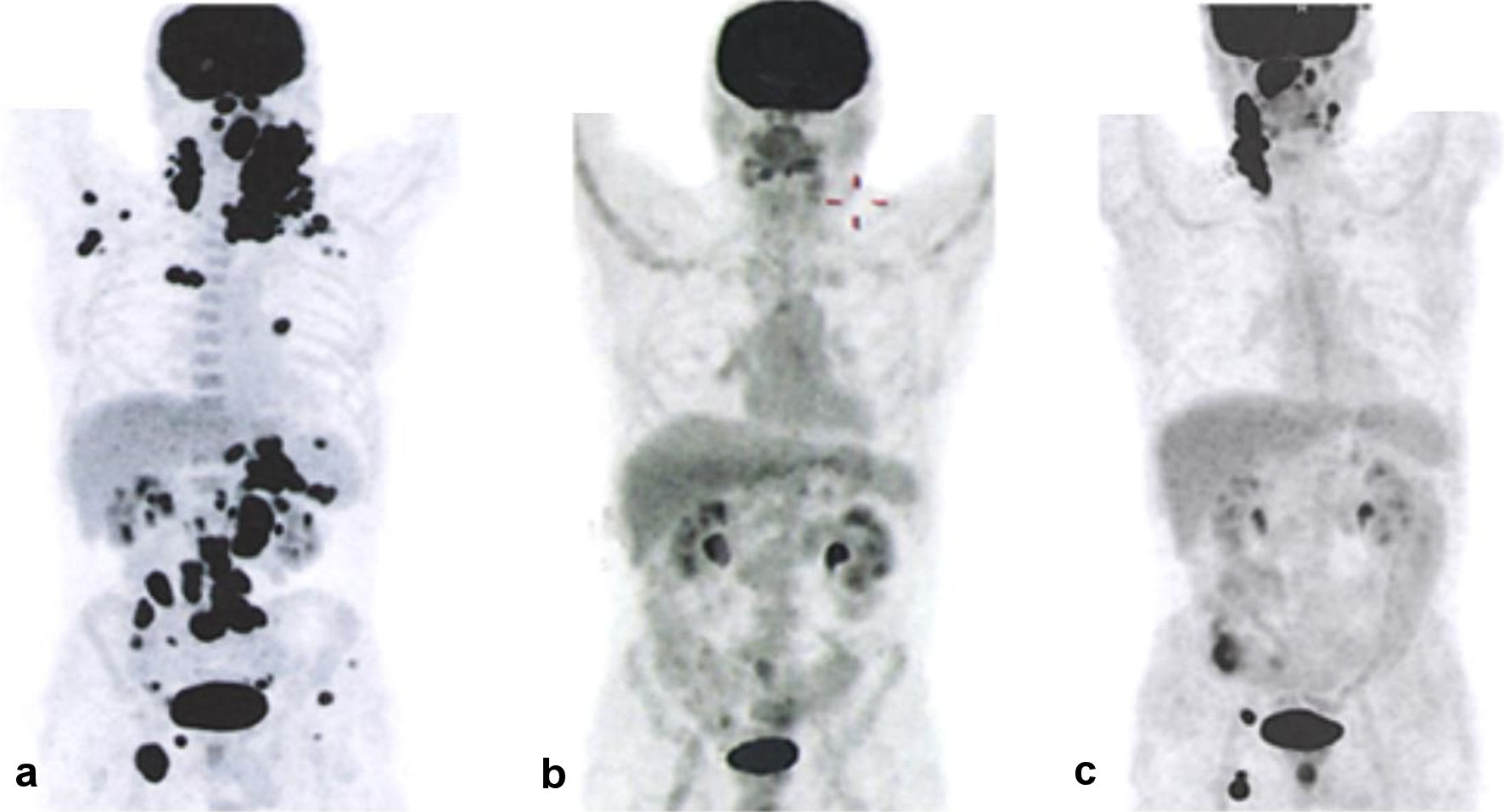 Click for large image | Figure 1. PET/CT results showing disease progression. (a) Before the start of therapy. (b) After the end of therapy. (c) Following disease relapse. PET/CT: positron emission tomography combined with computed tomography. |
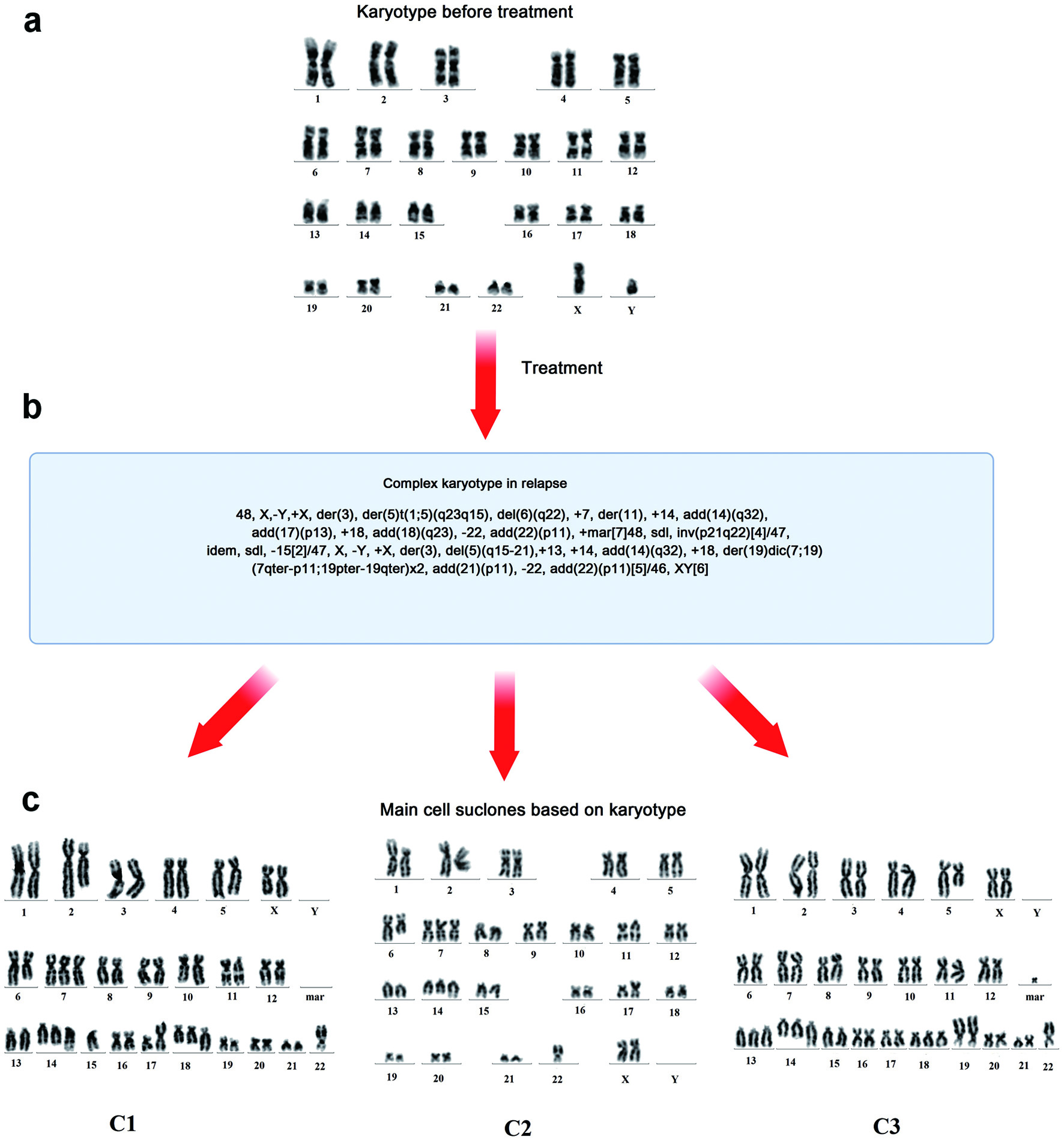 Click for large image | Figure 2. Results of cytogenetic examination of the cervical lymph node biopsy during disease onset and relapse. (a) Karyotype before treatment. (b) Karyotype after treatment. (c) Three main cell subclones after treatment and their karyotypes. c1: 48, X,-Y,+X,der(3),der(5)t(1;5)(q23q15), del(6)(q22),+7,der(11),+14,add(14)(q32), add(17(p13), +18,add(18)(q23),-22,add(22)(p11),+mar[7]48; c2: sdl,inv(p21q22)[4]/47,idem,sdl,-15[2]/47; c3: X,-Y,+X,der(3),del(5)(q15-21),+13,+14,add(14)(q32),+18,der(19)dic(7;19) (7qter→p11::19pter→19qter)×2,add(21)(p11),-22,add(22)(p11)[5]/46,XY[6]. der: derivative chromosome; del: deletion; add: additional; +mar: marker chromosome; sdl: sidelines; inv: inversion. |
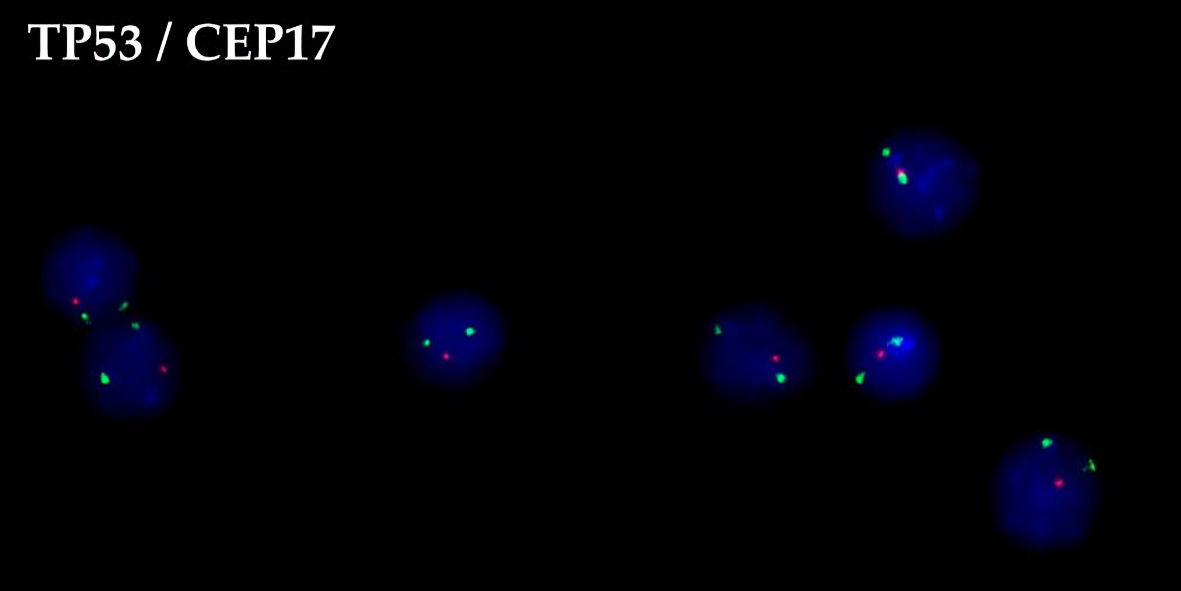 Click for large image | Figure 3. FISH of the 17p13 deletion in cervical lymph node biopsy following disease relapse. FISH: fluorescence in situ hybridization. |
In the course of further research, exome sequencing of the patient’s DNA was carried out on recurrent tumor and normal tissue (blood) samples. The data were then filtered according to the list of genes associated with oncological diseases of the lymphoproliferative system. As a result, 299 variations were identified, mostly single-nucleotide substitutions. A feature of this patient was the large number of mutations related to a single gene, five to eight mutations were observed in almost all analyzed genes.
The samples were then examined for the presence of pathogenic mutations. Thus, 141 mutations were identified, of all these mutations, only six are included in the dbSNP database, one of which is also in the ClinVar database, and the remaining 135 variants have not previously been discovered. Details of the mutations that were found are listed along with their predicted damaging effect here (Supplementary Material 1, www.wjon.org).
| Discussion | ▴Top |
According to the literature, mutations leading to DLBCL are found in a number of functionally significant genes. Among these genes are those whose functional role in the pathogenesis of DLBCL has been studied for many years, including CD79B, TP53, CARD11, MyD88, CREBBP, and EZH2 [8, 24-26]. To date, there are a number of other genes for which mutations were confirmed to have pathological significance for the development of DLBCL: MEF2B, MLL, BTG1, GNA13, ACTB, P2RY8, PCLO, TNFRSF14, KRAS, BRAF, BCL2, BCL6, MYC, TNFAP3, TCF4, and NOTCH1, and NOTCH2 [27-29].
The TP53 gene is an anti-oncogene, since the encoded p53 protein functions in preventing the formation of malignant tumors. Disruption of TP53 gene function is one of the most universal indicators of functional changes in the cells of various neoplasms. Activation of TP53 leads to changes in the expression level of more than 500 other genes. Deficiency in its function manifests as a loss of control over the cell cycle, apoptosis, aging processes, chromosomal stability, and excisional DNA repair. TP53 gene loss is accompanied by an uncontrolled accumulation of genetic damage that leads to malignant cell growth, and as a result, to the death of the body. Our patient had a TP53/17p13 deletion, which is associated with a particularly poor prognosis of the disease course and extremely low survival. Together with the 17p deletion, pathogenic mutations are often found in the second allele of the TP53 gene, leading to its inactivation. In the case studied here, no such mutations were found.
Multiple mutations of the KMT2A (MLL1)/KMT2D (MLL2) genes were also detected. The proteins encoded by these genes are lysine methyltransferases responsible for the methylation of lysine residues on histones, particularly histones H3 and H4 during oogenesis and early development [30]. Dysregulation of this methylation is crucial in the development of cancer. In the literature, numerous inactivating mutations have been described in medulloblastoma [31] and multiple myeloma [32], and chromosomal translocations involving a member of the MLL1 family are described in detail in acute leukemia [33]. In the tumor sample from our patient, we found seven mutations in the KMT2D gene and five mutations in the KMT2A gene that have not previously been described, and according to the results of predictor programs, have an inactivating effect.
The MEF2B gene encodes a transcription activator, and according to the literature, mutates in about 11% of cases of DLBCL, and about 12% of cases of follicular lymphomas. Ying et al in one of his papers showed that MEF2B can directly activate the transcription of the BCL6 protooncogene in normal B cells of the germinal center and is necessary for the proliferation of DLBCL [34]. Mutations enhance the transcriptional activity of MEF2B, either by disrupting its interaction with the co-repressor CABIN1, or by rendering it insensitive to mediated phosphorylation and sumoylation, which inhibit signal transmission. Consequently, the transcriptional activity of BCL6 is deregulated in DLBCL, with MEF2B mutations. Somatic mutations of MEF2B can lead to unregulated expression of the BCL6 oncogene in diffuse B-large cell lymphoma [34]. In our study, we found a non-synonymous substitution of p. L245M and a nonsense mutation of p. Q14*.
The CREBBP gene encodes a highly conserved and ubiquitously expressed nuclear phosphoprotein, which, together with the closely related EP300 protein, belongs to the KAT3 family of histone/protein lysine acetyltransferases. By modifying lysine residues on both histone and non-histone nuclear proteins, CREBBP and EP300 function as transcriptional coactivators for a large number of DNA-binding transcription factors involved in multiple signaling and developmental pathways [35, 36]. About 39% of cases of DLBCL and 41% of cases of follicular lymphoma show genomic deletions and/or somatic mutations that remove or inactivate the HAT coding domain of these two genes. In addition, it was demonstrated that the mutant CREBBP and EP300 proteins are deficient in the acetylation of BCL6 and p53, which leads to constitutive activation of the oncoprotein and a decrease in the activity of the tumor suppressor p53 [37]. In our study, the inactivating mutation p.M1385I in the HAT domain of the CREBBP gene and the nonsense mutation p.C380* were detected, but no mutations were found in the EP300 gene.
EZH2 is a catalytic subunit of polycomb repressive complex 2 (PRC2), which along with other components of PRC2, suppresses gene expression by methylation of histone H3 in lysine 27. EZH2 aberration is observed in a wide range of cancers, including several categories of B-cell and T-cell malignancies, and is associated with a poor clinical prognosis and outcomes. EZH2 plays a significant role in the development of the lymphoid system, so its deregulation, due to genetic or non-genetic causes, contributes to the development of lymphoma or leukemia associated with B-cells as well as T-cells [38, 39]. We identified two non-synonymous mutations in the EZH2 gene, p. Q730H and p.K492N, as well as four mutations with a reading frame shift. The effect of these mutations on protein function is unknown.
The protooncogene BCL6 has been shown to be deregulated due to t(3; 14) (q27; q32) translocation in 5-10% of DLBCL cases [40]. In addition, somatic mutations in BCL6 that disrupt the negative autoregulation of its expression have been reported in cases of DLBCL [41]. In our case, we observed not only a translocation involving loci of the BCL6/3q27 gene, but also three mutations in this gene with pathogenic significance: p. P483T, p.P286S, and p.C475*.
Translocation of BCL2 t(14;18), detected in 20% of DLBCL cases [40], leads to an increase in the activity of the BCL2 oncogene, and as a result, an increase in the survival of tumor cells. We detected this rearrangement in our patient, but there were no point mutations of pathogenic value in the BCL2 gene.
MYC is a protooncogene encoding the Myc protein, a transcription factor of great importance for proliferation, metabolism, differentiation, apoptosis, microenvironment remodeling, and immune responses. MYC deregulation has oncogenic potential, which leads to increased cell proliferation, angiogenesis, apoptosis, genome instability, and inhibition of cell differentiation [42]. In addition to Burkitt’s lymphoma, MYC gene rearrangements were detected in 5-15% of patients with DLBCL as well as in other types of lymphomas [43, 44]. In patients with DLBCL, MYC rearrangement has been shown to be associated with worse survival prognosis [45]. According to various literature data, it is known that mutations in the MYC gene in DLBCL are not as well studied as translocations. Xu-Monette et al found a wide range of single-nucleotide variations of MYC gene in DLBCL that have different functional and clinical effects [46]. In this study, we did not detect any rearrangements of the MYC gene in the patient, but we found four single-nucleotide variations with pathogenic significance, namely p. E400*, p.E399*, p.E385*, and p.P43L, although the clinical effect these mutations is unknown.
According to the conducted studies, more than 50% of ABC-DLBCL and a small proportion of GCB-DLBCL cases involve somatic mutations in several genes of the NF-κB signaling pathway that regulates signal transmission. The TNFAIP3 gene, which encodes the ubiquitin-modifying enzyme (A20) involved in terminating NF-κB responses, is frequently affected. At the same time, missense mutations of CARD11 produce molecules with a significantly increased ability to activate NF-κB pathway. Thus, the regulation of the NF-κB pathway in DLBCL caused by genetic changes in these genes that lead to their loss or activation may contribute to lymphomagenesis [24, 47]. In our analysis, we found mutations in both the CARD11 and TNFAIP3 genes.
ATM encodes a serine/threonine kinase belonging to the phosphatidylinositol-3-kinase (PI3-K) family and plays a central role in signaling pathways activated by DNA damage. ATM kinase normally plays a key role in the response to DNA damage, being a tumor suppressor gene and part of the ATM-p53 pathway. DNA damage induces activation of ATM and ATR kinases, which, together with the p53 and p14/ARF proteins, can induce cell cycle arrest, DNA repair, or apoptosis. In patients with DLBCL, ATM mutations were shown to be associated with inactivation of the ARF tumor suppressor gene and the P53 tumor protein (TP53) [48]. It has been suggested that mutations in ATM and inactivation of the ARF-TP53 pathway may contribute to the development of DLBCL. Several studies have shown that ATM may be involved in the development of certain subtypes of sporadic lymphoma and leukemia: missense mutations and loss of function in the ATM gene have been demonstrated in T-cell prolymphocytic leukemia [49], mantle cell lymphoma [50], B-cell chronic lymphocytic leukemia [51], diffuse B-large cell lymphoma, and follicular lymphoma [48]. The protein kinase encoded by the ATM gene is involved in regulation of the activity of the p53 protein encoded by the TP53 gene. P53 activity may decrease when the function of the ATM gene is lost. In our case, nine mutations with a poor prognosis were detected in the ATM gene.
The protein encoded by the ATR gene is a serine/threonine protein kinase, a key activator of cellular responses to DNA damage caused by ultraviolet light, ionizing radiation, and halting of DNA replication. In response to these events, ATR phosphorylates key proteins in various branches of the DNA damage response pathways, such as p53, BRCA1, CHK1, and Rad17, thereby activating DNA repair, cell cycle checkpoints, or apoptosis [52]. An important role of ATR mutations in hereditary predisposition to breast and ovarian cancer has been demonstrated [53]. One paper shows the significance of ATM and ATR mutations for the initiation and progression of pyothorax-related lymphoma [54]. Using exome sequencing, seven mutations with pathogenic significance were detected in the ATR gene.
Phosphatidylinositol-3-kinases (PI3Ks) are lipid kinases that regulate signaling pathways important for neoplasia, including cell proliferation, adhesion, survival, and motility. We found pathogenic mutations in the PIK3CA and PIK3CD genes of our patient. The large number of mutations observed in these genes indicates their functional importance, and the dysregulation of the PI3 kinase pathway is a common feature of many cancers [55, 56]. In addition, mutations have been identified in the MTOR gene, one of the known oncogenes in the PI3 kinase pathway, which also indicates deregulation of the PI3 kinase pathway [57]. In our work, we found three nonsynonymous substitutions that probably lead to damage to the protein encoded by the MTOR gene.
We identified five protein-damaging mutations in the ALK gene encoding anaplastic lymphoma kinase. This kinase was first detected in anaplastic large cell lymphoma as a result of translocation between chromosomes (2; 5) (p23: q35). Changes in the ALK gene are implicated in neuroblastoma, lung cancer, and other malignancies [58, 59].
GTPases of the RHO family are guanine-nucleotide-binding enzymes that bind guanosine triphosphate (GTP) and catalyze its hydrolysis to guanosine diphosphate (GDP). RHO GTPases are active in the GTP-bound state and inactive in the GDP-bound state, and the relationship between GTP/GDP-bound conformations (active/inactive) is critical for proper intracellular signaling [60]. The RHO GTPase family consists of 18 to 22 members. They can be classified according to their homology and structure into the following subfamilies: typical and atypical. Both typical and atypical RHO GTPases are critical transducers of intracellular signaling and are associated with human cancers. Moreover, both gain of function and loss of function mutations have been described in various tumors [61]. Studies using whole exome sequencing technology have identified repeated RHOA mutations in various human lymphomas of both B-cell and T-cell origin, including diffuse B-large cell lymphoma [62]. Further studies of the biological significance of these mutations suggested a leading role for RHOA in the pathogenesis of these lymphomas. In our patient, three non-synonymous substitutions with a pathogenic value in the RHOA gene were identified: p. P101T, p.A177D, and p.W99C.
Proteins of the NOTCH family are integral receptor proteins, represented in humans by four types (NOTCH 1 - 4). These proteins control the proliferation, differentiation, and development of cells and tissues, as well as activate the transcription of genes involved in regulating the balance between these processes [63]. In adult tissues, NOTCH-mediated signals are important regulators in maintaining self-renewal, promoting, for example, myogenesis, neurogenesis, and lymphocyte development [64]. Given the important role of NOTCH proteins in a wide range of processes and tissues, aberrations leading to an increase or loss of NOTCH signaling components and functions are associated with a variety of disorders, including solid cancer and hematological malignancies, where NOTCH can act as an oncogene or as a tumor suppressor. Currently, there are many examples that clearly show the relationship of mutations in the Notch1 and Notch2 genes with lymphoproliferative disorders, including chronic lymphocytic leukemia, mantle lymphoma, marginal spleen lymphoma, diffuse B-large cell lymphoma, follicular lymphoma, and Burkitt and Hodgkin lymphomas [65-68]. According to our data, the patient has an inactivating nonsense mutation p.Y1508* in their NOTCH2 gene as well as a non-synonymous replacement of p.G736V with a pathogenic value.
In addition, pathogenic mutations with a damaging effect were detected in a number of other genes, namely BCL10, BCL9, CD58, XPO1, DTX3L, PARP14, TP63, MAP3K1, IRF4, CCND3, RAD54B, NFKB2, STAT6, DDX3X, MLLT10, DNTT, and BCR, which are associated with different types of lymphomas. Recently, genomic studies have focused on the identification of genetic subtypes of DLBCL, which are dictated by the genetic, phenotypic and clinical heterogeneity of this type of lymphoma, and as a result, a different response to generally accepted treatment regimens [21].
Conclusions
Our clinical case is characterized by an atypical picture of the disease course, where the primary chemotherapy regimen gave satisfactory results. However, after 2 months, the patient experienced rapid relapse, and as a result, their clinical outlook deteriorated significantly. In cytogenetic studies, multiple focal formations and significant chromosome breakdowns were observed. We found, in our patient, mutations that affect components of signaling pathways such as NOTCH2 and NF-κB, a number of epigenetic regulators (KMT2D, CREBBP, EP300, ARID1A, MEF2B), as well as members of the immunoglobulin receptor superfamily (CD58 and CD70). Without data on the primary tumor, it is impossible to say with certainty whether these mutations were the result of therapy or were originally present in the tumor. Possibly, if similar data had been available when making the diagnosis and developing the treatment regimen, a different tactic would have been chosen. The importance of research and development in the field of molecular therapy for DLBCL cannot be overemphasized. We believe that future research should focus on an in-depth search for predictive biomarkers based on genomic, transcriptomic and epigenetic data and on the development of molecular tools and delivery systems to target tissues and organs. In addition, pharmacogenetic studies that anticipate the effects of specific therapy and studies related to the effects on the molecular pathways of anticancer drugs (such as lenalidomide during remission and relapse) are promising.
| Supplementary Material | ▴Top |
Suppl 1. Somatic mutations detected in tumor biopsies.
Acknowledgments
None to declare.
Financial Disclosure
This research was funded by an internal grant of the National Research Center Kurchatov Institute #1360 of 25.06.2019 (development of markers to identify different types of diffuse large B-cell lymphoma (DLBCL)). The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
Informed consent was obtained from the patient. All patient information was deidentified.
Author Contributions
Conceptualization: EZ, S. Tsygankova, MGA and NG; methodology: D. Komova., EB, NS, S. Tsygankova; software: FS and SR; validation: D. Komova., EB, NS, AS, TO and S. Tsygankova; formal analysis: FS and SR; investigation: D. Komova, D. Koroleva, NG, EB, NS, AS, S. Tatarnikova, TO and S. Tsygankova; resources: S. Tsygankova and EZ; data curation: FS; writing-original draft preparation: NG, S. Tsygankova, and AN; writing-review and editing: MGA, NG, S. Tsygankova, EZ, and AN; visualization: D. Koroleva, NG, and SR; supervision: S. Tsygankova and EZ; project administration: S. Tsygankova and EZ; funding acquisition: S. Tsygankova. All authors have read and agreed to the published version of the manuscript.
Data Availability
All DNA sequences were uploaded to the NCBI Sequence Read Archive (BioProject ID PRJNA752256).
Abbreviations
DLBCL: diffuse large B-cell lymphoma; CHOP: cyclophosphamide, hydroxydaunorubicin, hydrochloride, vincristine, prednisone; R-CHOP: rituximab, cyclophosphamide, hydroxydaunorubicin, hydrochloride, vincristine, prednisone; auto-SCT: autologous stem cell transplantation; ABC-DLBCL: activated B-cell subtype of diffuse large B-cell lymphoma; auto-HSCT: autologous hematopoietic stem cell transplantation; CT: chemotherapy; FISH: fluorescent in situ hybridization; DHL: double-hit lymphoma; IPI: international Prognostic Index; NPS: non-progressive survival; NGS: next-generation sequencing; WES: whole exome sequencing; non-GCB: non-germinal center B-cell; PET/CT: positron emission tomography combined with computed tomography; LDH: lactate dehydrogenase; SCE: standard cytogenetic examination
| References | ▴Top |
- Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, Morel P, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235-242.
doi pubmed - Visco C, Li Y, Xu-Monette ZY, Miranda RN, Green TM, Li Y, Tzankov A, et al. Comprehensive gene expression profiling and immunohistochemical studies support application of immunophenotypic algorithm for molecular subtype classification in diffuse large B-cell lymphoma: a report from the International DLBCL Rituximab-CHOP Consortium Program Study. Leukemia. 2012;26(9):2103-2113.
doi pubmed - Vitolo U, Trneny M, Belada D, Burke JM, Carella AM, Chua N, Abrisqueta P, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol. 2017;35(31):3529-3537.
doi pubmed - Rastorguev S, Koroleva D, Boulygina E, et al. Clinical and prognostic value of molecular markers of diffuse large B-cell lymphoma. Clin Oncohematology. 2018;12(1):95-100.
doi - Sesques P, Johnson NA. Approach to the diagnosis and treatment of high-grade B-cell lymphomas with MYC and BCL2 and/or BCL6 rearrangements. Blood. 2017;129(3):280-288.
doi pubmed - Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503-511.
doi pubmed - Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(25):1937-1947.
doi pubmed - Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001;194(12):1861-1874.
doi pubmed - Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, Kohlhammer H, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88-92.
doi pubmed - Mondello P, Steiner N, Willenbacher W, Ferrero S, Ghione P, Marabese A, Pitini V, et al. Lenalidomide in Relapsed or Refractory Diffuse Large B-Cell Lymphoma: Is It a Valid Treatment Option? Oncologist. 2016;21(9):1107-1112.
doi pubmed - Nowakowski GS, LaPlant B, Macon WR, Reeder CB, Foran JM, Nelson GD, Thompson CA, et al. Lenalidomide combined with R-CHOP overcomes negative prognostic impact of non-germinal center B-cell phenotype in newly diagnosed diffuse large B-Cell lymphoma: a phase II study. J Clin Oncol. 2015;33(3):251-257.
doi pubmed - Jimenez-Cortegana C, Palazon-Carrion N, Martin Garcia-Sancho A, Nogales-Fernandez E, Carnicero-Gonzalez F, Rios-Herranz E, de la Cruz-Vicente F, et al. Circulating myeloid-derived suppressor cells and regulatory T cells as immunological biomarkers in refractory/relapsed diffuse large B-cell lymphoma: translational results from the R2-GDP-GOTEL trial. J Immunother Cancer. 2021;9(6):e002323.
doi pubmed - Gascoyne DM, Banham AH. The significance of FOXP1 in diffuse large B-cell lymphoma. Leuk Lymphoma. 2017;58(5):1037-1051.
doi pubmed - Ryan RJ, Nitta M, Borger D, Zukerberg LR, Ferry JA, Harris NL, Iafrate AJ, et al. EZH2 codon 641 mutations are common in BCL2-rearranged germinal center B cell lymphomas. PLoS One. 2011;6(12):e28585.
doi pubmed - Young KH, Leroy K, Moller MB, Colleoni GW, Sanchez-Beato M, Kerbauy FR, Haioun C, et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: an international collaborative study. Blood. 2008;112(8):3088-3098.
doi pubmed - Yu X, Li W, Deng Q, Li L, Hsi ED, Young KH, Zhang M, et al. MYD88 L265P mutation in lymphoid malignancies. Cancer Res. 2018;78(10):2457-2462.
doi pubmed - Zenz T, Kreuz M, Fuge M, Klapper W, Horn H, Staiger AM, Winter D, et al. TP53 mutation and survival in aggressive B cell lymphoma. Int J Cancer. 2017;141(7):1381-1388.
doi pubmed - Zhao H, Kan Y, Wang X, Chen L, Ge P, Qian Z. Genetic polymorphism and transcriptional regulation of CREBBP gene in patient with diffuse large B-cell lymphoma. Biosci Rep. 2019;39(8):BSR20191162.
doi pubmed - Bojarczuk K, Chapuy B. Molecular classification of aggressive B-cell lymphoma. HemaSphere. 2019;3(S2):116-118.
doi - Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, Lawrence MS, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24(5):679-690.
doi pubmed - Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, Wang JQ, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell. 2020;37(4):551-568.e514.
doi pubmed - Zvonkov EE, Morozova AK, Kravchenko SK. Treatment of adult patients with primary diffuse large B-lymphosarcoma of stomach on the modified program NHL-BFM-90. Treatment of diseases of the blood system. Published online. 2012:659-677.
- Gabeeva NG, Zvonkov EE, Koroleva DA, Chukavina MM, Obukhova TN, Kovrigina AM. Successful experience of treatment of a patient with generalized non-GCB- DLBCL using the R-mNHL-BFM-90 protocol with lenalidomide: case report and review of literature. Ter Arkh. 2018;90(7):96-101.
doi pubmed - Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, Dave SS, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319(5870):1676-1679.
doi pubmed - Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181-185.
doi pubmed - Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115-119.
doi pubmed - Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109(10):3879-3884.
doi pubmed - Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298-303.
doi pubmed - Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, Wells VA, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43(9):830-837.
doi pubmed - Andreu-Vieyra CV, Chen R, Agno JE, Glaser S, Anastassiadis K, Stewart AF, Matzuk MM. MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol. 2010;8(8):e1000453.
doi pubmed - Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331(6016):435-439.
doi pubmed - Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467-472.
doi pubmed - Coenen EA, Raimondi SC, Harbott J, Zimmermann M, Alonzo TA, Auvrignon A, Beverloo HB, et al. Prognostic significance of additional cytogenetic aberrations in 733 de novo pediatric 11q23/MLL-rearranged AML patients: results of an international study. Blood. 2011;117(26):7102-7111.
doi pubmed - Ying CY, Dominguez-Sola D, Fabi M, Lorenz IC, Hussein S, Bansal M, Califano A, et al. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol. 2013;14(10):1084-1092.
doi pubmed - Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14(13):1553-1577.
doi - Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87(5):953-959.
doi - Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471(7337):189-195.
doi pubmed - Li B, Chng WJ. EZH2 abnormalities in lymphoid malignancies: underlying mechanisms and therapeutic implications. J Hematol Oncol. 2019;12(1):118.
doi pubmed - Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109(52):21360-21365.
doi pubmed - Willis TG, Dyer MJ. The role of immunoglobulin translocations in the pathogenesis of B-cell malignancies. Blood. 2000;96(3):808-822.
doi - Pasqualucci L, Migliazza A, Basso K, Houldsworth J, Chaganti RS, Dalla-Favera R. Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood. 2003;101(8):2914-2923.
doi pubmed - Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008;8(12):976-990.
doi pubmed - Hu Z, Medeiros LJ, Chen Z, Chen W, Li S, Konoplev SN, Lu X, et al. Mantle cell lymphoma with MYC rearrangement: a report of 17 patients. Am J Surg Pathol. 2017;41(2):216-224.
doi pubmed - Wang XJ, Medeiros LJ, Bueso-Ramos CE, Tang G, Wang S, Oki Y, Desai P, et al. P53 expression correlates with poorer survival and augments the negative prognostic effect of MYC rearrangement, expression or concurrent MYC/BCL2 expression in diffuse large B-cell lymphoma. Mod Pathol. 2017;30(2):194-203.
doi pubmed - Li S, Weiss VL, Wang XJ, Desai PA, Hu S, Yin CC, Tang G, et al. High-grade B-cell lymphoma with MYC rearrangement and without BCL2 and BCL6 rearrangements is associated with high P53 expression and a poor prognosis. Am J Surg Pathol. 2016;40(2):253-261.
doi pubmed - Xu-Monette ZY, Deng Q, Manyam GC, Tzankov A, Li L, Xia Y, Wang XX, et al. Clinical and biologic significance of MYC genetic mutations in de novo diffuse large B-cell lymphoma. Clin Cancer Res. 2016;22(14):3593-3605.
doi pubmed - Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459(7247):717-721.
doi pubmed - Gronbaek K, Worm J, Ralfkiaer E, Ahrenkiel V, Hokland P, Guldberg P. ATM mutations are associated with inactivation of the ARF-TP53 tumor suppressor pathway in diffuse large B-cell lymphoma. Blood. 2002;100(4):1430-1437.
doi pubmed - Stoppa-Lyonnet D, Soulier J, Lauge A, Dastot H, Garand R, Sigaux F, Stern MH. Inactivation of the ATM gene in T-cell prolymphocytic leukemias. Blood. 1998;91(10):3920-3926.
doi pubmed - Schaffner C, Idler I, Stilgenbauer S, Dohner H, Lichter P. Mantle cell lymphoma is characterized by inactivation of the ATM gene. Proc Natl Acad Sci U S A. 2000;97(6):2773-2778.
doi pubmed - Bullrich F, Rasio D, Kitada S, Starostik P, Kipps T, Keating M, Albitar M, et al. ATM mutations in B-cell chronic lymphocytic leukemia. Cancer Res. 1999;59(1):24-27.
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300(5625):1542-1548.
doi pubmed - Heikkinen K, Mansikka V, Karppinen SM, Rapakko K, Winqvist R. Mutation analysis of the ATR gene in breast and ovarian cancer families. Breast Cancer Res. 2005;7(4):R495-501.
doi pubmed - Liu A, Takakuwa T, Fujita S, Ham MF, Luo WJ, Daibata M, Aozasa K. Alterations of DNA damage-response genes ATM and ATR in pyothorax-associated lymphoma. Lab Invest. 2005;85(3):436-446.
doi pubmed - Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Br J Cancer. 2006;94(4):455-459.
doi pubmed - Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554.
doi pubmed - Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, Dunphy C, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110(4):1398-1403.
doi pubmed - Carneiro BA, Pamarthy S, Shah AN, Sagar V, Unno K, Han H, Yang XJ, et al. Anaplastic lymphoma kinase mutation (ALK F1174C) in small cell carcinoma of the prostate and molecular response to alectinib. Clin Cancer Res. 2018;24(12):2732-2739.
doi pubmed - Morris SW, Naeve C, Mathew P, James PL, Kirstein MN, Cui X, Witte DP. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin's lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK). Oncogene. 1997;14(18):2175-2188.
doi pubmed - Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol. 2016;17(8):496-510.
doi pubmed - Bustelo XR. RHO GTPases in cancer: known facts, open questions, and therapeutic challenges. Biochem Soc Trans. 2018;46(3):741-760.
doi pubmed - Voena C, Chiarle R. RHO family GTPases in the biology of lymphoma. Cells. 2019;8(7):646.
doi pubmed - Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216-233.
doi pubmed - Tanigaki K, Kuroda K, Han H, Honjo T. Regulation of B cell development by Notch/RBP-J signaling. Semin Immunol. 2003;15(2):113-119.
doi - Kiel MJ, Velusamy T, Betz BL, Zhao L, Weigelin HG, Chiang MY, Huebner-Chan DR, et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med. 2012;209(9):1553-1565.
doi pubmed - Vaisitti T, Arruga F, Deaglio S. Targeting the adenosinergic axis in chronic lymphocytic leukemia: a way to disrupt the tumor niche? Int J Mol Sci. 2018;19(4):1167.
doi pubmed - Xiu MX, Liu YM. The role of oncogenic Notch2 signaling in cancer: a novel therapeutic target. Am J Cancer Res. 2019;9(5):837-854.
- Zhang X, Shi Y, Weng Y, Lai Q, Luo T, Zhao J, Ren G, et al. The truncate mutation of Notch2 enhances cell proliferation through activating the NF-kappaB signal pathway in the diffuse large B-cell lymphomas. PLoS One. 2014;9(10):e108747.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.