

| World Journal of Oncology, ISSN 1920-4531 print, 1920-454X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, World J Oncol and Elmer Press Inc |
| Journal website https://www.wjon.org |
Original Article
Volume 14, Number 6, December 2023, pages 488-498

Prevalence and Prognosis of Secondary Genetic Aberrations Among Patients With Core Binding Factor Acute Myeloid Leukemia: A Mitelman Database Analysis
Renzo Martin Chapilliquen Ramireza, b ![]() , Mariana Teresa de Jesus Corbacho Pachasa
, Mariana Teresa de Jesus Corbacho Pachasa ![]() , Richard Junior Zapata Dongoa
, Richard Junior Zapata Dongoa ![]()
aFacultad de Medicina Humana, Universidad de Piura, Miraflores 15074, Lima, Peru
bCorresponding Author: Renzo Martin Chapilliquen Ramirez, Facultad de Medicina Humana, Universidad de Piura, Miraflores 15074, Lima, Peru
Manuscript submitted August 27, 2023, accepted October 24, 2023, published online November 18, 2023
Short title: Secondary Genetic Aberrations in CBF-AML
doi: https://doi.org/10.14740/wjon1661
| Abstract | ▴Top |
Background: Core binding factor acute myeloid leukemia (CBF-AML) comprises t(8;21) and inv(16) and usually has a favorable prognosis. However, a wide spectrum of secondary genetic aberrations has been shown to be associated with worse outcomes with respect to overall survival (OS) and relapse. We aimed to identify secondary molecular and chromosomal aberrations within each group of CBF-AML, i.e., t(8;21) and inv(16), and to evaluate their prognosis with OS.
Methods: Using the Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer, we analyzed 193 cases of CBF-AML reported between 2011 and 2021. We conducted a survival analysis to determine the 5-year OS, and we conducted univariate and multivariate Cox regression to identify independent genetic factors related to OS.
Results: Among the 193 cases with CBF-AML, structural and numerical chromosome rearrangements were 25.9% and 40.9%, respectively, and secondary genetic mutations were 54.9%. The 5-year OS for the presence of del(7) and trisomy 22 was significantly worse. NRAS mutations had a worse 5-year OS in the t(8;21) group in the univariate analysis but showed no significant difference in the multivariate analysis.
Conclusions: CBF-AML has heterogeneous cytogenetic characteristics but no difference in the 5-year OS between the inv(16) and t(8;21) groups. Finally, the presence of del(7), trisomy 22 and NRAS mutations showed a potential prognostic impact in CBF-AML patients. Secondary genetic findings may need to be identified to determine its association to a worse prognosis, and in the future develop better targeted therapies in patients with CBF-AML.
Keywords: Leukemia; Myeloid; Acute; Core binding factors; Chromosome aberrations; Oncogenes; Prognosis
| Introduction | ▴Top |
Acute myeloid leukemia (AML) is a common and heterogeneous hematological malignancy with approximately 20,000 new cases per year in the United States [1]. In recent years, the classification of AML has evolved based on cytogenetic and molecular analyses, which establish the risk category and guide management [2]. The most frequent subtypes of AML include AML with t(8;21)(q22;q22) and AML with inv(16)(p13.1;q22), also known as core binding factor acute myeloid leukemia (CBF-AML) [3].
Core binding factor (CBF) is a heterodimeric transcription factor that regulates leukemogenesis and consists of RUNX1 and CBFβ subunits [4]. In CBF-AML, t(8;21) and inv(16) rearrangements alter these subunits and form the fusion genes RUNX1-RUNX1T1 and CBFβ-MYH11, respectively, which block myeloid differentiation. However, experimental studies have demonstrated that additional genetic mutations are needed to initiate leukemogenic transformation [5].
CBF-AML accounts for approximately 30% and 15% of pediatric and adult AML cases, respectively [6]. CBF with inv(16) accounts for approximately 8% of AML cases and is predominant in adult patients, while t(8;21) accounts for approximately 5% of AML cases and is more frequent among younger individuals [5]. According to the World Health Organization (WHO) 2008 classification, CBF-AML has a favorable prognosis, with a 5-year overall survival (OS) of 50% and complete remission (CR) rates of 88% [7, 8]. Nevertheless, 50% of patients relapse due to secondary molecular or chromosomal aberrations that have been found in approximately 52% of CBF-AML patients [7, 9].
The predominant additional chromosome abnormality found in CBF with t(8;21) is the loss of sex chromosomes, while in inv(16) is trisomy 22 [9, 10]. The most common molecular mutations found in CBF-AML are in the KIT, FLT3, TET2 and RAS genes [9]. KIT mutation is associated with an adverse prognosis with the lowest OS rate and higher risk of relapse, especially in t(8;21) [11].
The wide diversity in secondary aberrations has led to persistent challenges in defining the prognosis and establishing effective therapies in CBF-AML. Additionally, there is controversy regarding group t(8;21) and inv(16) in CBF-AML due to clinical, pathological, and molecular heterogeneity [8]. In this secondary database study, we aimed to identify the prevalence of secondary molecular and chromosomal aberrations within each group of CBF-AML, i.e., t(8;21) and inv(16), and to evaluate their prognosis by 5-year OS analysis.
| Materials and Methods | ▴Top |
Study design and data sources
We conducted an observational and transversal study of secondary databases. The National Cancer Institute’s Mitelman Database of Chromosome Aberrations and Gene Fusions was used to identify patients with CBF-AML. Each patient had a source study from which the data were collected. Finally, a new database was created in Excel and then cleaned up in RStudio.
Participants
On the date of inquiry (March 21, 2022), 2,947 cases with CBF-AML were obtained. Only cases published between 2010 and 2021 were included, leaving 411 cases. After the merger of duplicated cases because of relapse, 398 cases remained, of which 205 cases with missing data on relapse and OS were excluded. Ultimately, 193 cases were included in the current analysis (Supplementary Material 1, www.wjon.org).
Variables
The variables included for this study were CBF-AML group, sex, age at diagnosis, age group, French American British (FAB) classification, karyotype, number of chromosomes, structural rearrangements, numerical chromosome abnormalities, secondary genetic mutations, relapse, status, and 5-year OS, which was the main outcome. A detailed glossary was made for the variables included in the study, and all structural rearrangements, numerical chromosomal abnormalities, and secondary genetic mutations are specified there (Supplementary Material 2, www.wjon.org).
Statistical analysis
The statistical analysis was performed using the programming language R, version 4.1.2, and the integrated development environment RStudio 2021.09.1 + 372. Continuous variables are presented as medians with interquartile ranges (IQRs), and categorical variables are presented as absolute frequencies and percentages. Significance was determined by the Chi-squared test, Wilcoxon rank sum test and Fisher’s exact test, where P < 0.05 was considered statistically significant.
OS was defined as the length of time from diagnosis to death from any cause, based on available data from publications. Survival analysis was conducted for 5-year OS, and the differences between CBF-AML groups were described using Kaplan-Meier curves and compared by log-rank tests. Univariate Cox regression analyses were performed on 5-year OS for the entire sample and for the inv(16) and t(8;21) groups separately, so we ended with three models per variable. Multivariable Cox analysis included sex, age group and relapse. The analysis to identify genetic factors related to OS was performed for secondary genetic aberrations in general and for each variable separately. Kaplan-Meier curves of each aberration with a reduced frequency will not be presented with their confidence interval (CI) because of their amplitude.
Ethical considerations
This research did not involve human participation directly or biological samples. The data obtained from the reference studies of the cases registered in the Mitelman Database do not contain any information capable of identifying the included patients. This study was approved by the Institutional Ethics Committee in Research of University of Piura in September 2021, and conducted in compliance with the ethical standards of the responsible institution on human subjects as well as with the Helsinki Declaration.
| Results | ▴Top |
Data from 193 cases with CBF-AML were extracted and analyzed from papers published between 2010 and 2021. We classified the study design into case reports, case series and cohort-like studies. The latter included cohort studies or studies based on patient samples but with the clinical data needed for our analysis. We found 38 cases (19.7%) from case report studies, 19 cases in both the inv(16) and t(8;21) groups. There were four cases (2.1%) in case series papers, one case in the inv(16) group and three cases in the t(8;21) group. The greatest number of cases were from cohort-like studies, with 151 cases (78.2%), 56 in the inv(16) group and 95 in the t(8;21).
There was a predominance of males (n = 120, 62.2%), with a median age at diagnosis of 43.0 years (IQR: 25.0 - 53.0). In regard to FAB classification, the majority was AML not otherwise specified (NOS) (n = 128, 66.3%). Nevertheless, the second category with more cases was different in each group of CBF-AML. In the inv(16) group, the most common category was acute myelomonocytic leukemia (M4) (n = 18, 23.7%), and in the t(8;21) group, the most common category was AML with maturation (M2) (n = 30, 25.6%) (Table 1).
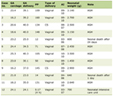 Click to view | Table 1. Characteristics of 193 CBF-AML Cases From Mitelman Database Between 2010 and 2021 |
We found 50 cases (25.9%) with structural chromosomal rearrangements, (9;22) and del(9), with total predominance in the inv(16) and t(8;21) groups, respectively. Approximately 60% of the cases had 46 chromosomes (n = 112), and there was a significant difference in the categories (< 46, 46 and > 46 chromosomes) between the CBF-AML groups (P < 0.001). Numerical chromosome abnormalities were present in 79 cases (40.9%), and loss of sex chromosomes (X and Y) was exclusive to the t(8;21) group and trisomy 8 and 22 in the inv(16) group, except for one case with t(8;21) who had trisomy 8. A summary of all chromosomal alterations can be found here (Supplementary Material 3, 4 www.wjon.org).
Secondary genetic mutations were present in 106 cases (54.9%), where we identified four gene fusions: PML-RARα, BCR-ABL1 and the CBF-AML rearrangements (MYH11-CBFB, RUNX1-RUNX1T1) (Fig. 1). We found another 45 mutations, but 30 of them were not recurrent. The BCRL-ABL1 and KIT mutations were predominant at inv(16) and BRCC3, CCND2, ASXL1 and ASXL2 at t(8;21). A summary of all secondary genetic mutations can be found here (Supplementary Material 5, www.wjon.org).
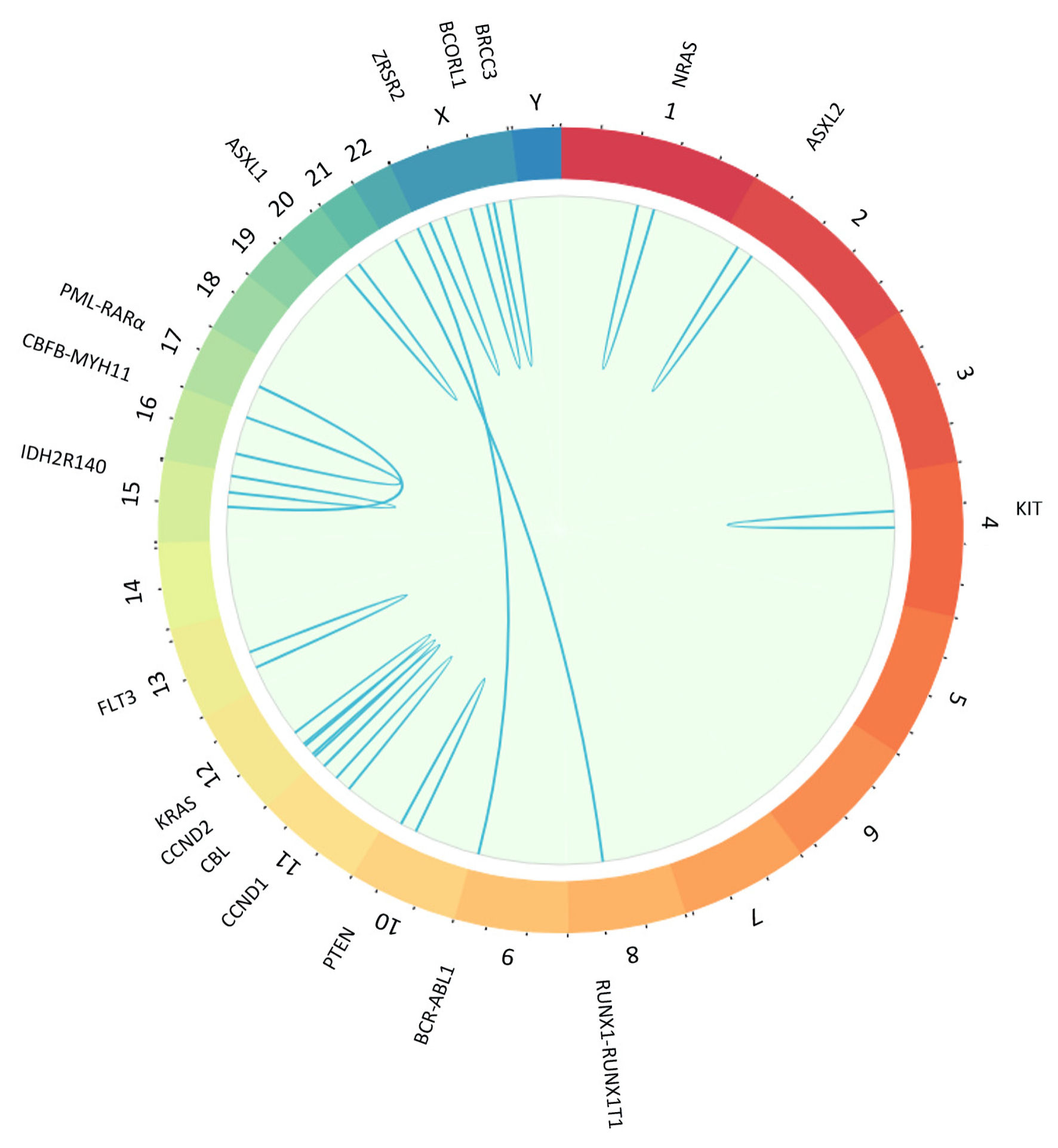 Click for large image | Figure 1. Plot created with BioCircos package in R showing with sky-blue lines secondary genetic mutations and gene fusions found in core binding factor acute myeloid leukemia (CBF-AML) cases in Mitelman Database. Chromosomes are individually colored and arranged clockwise from chromosome 1 to Y. We showed only mutations with a frequency greater than 1, and there were no rearrangements involving the Y chromosome. NRAS: neuroblastoma RAS viral gene mutation; ASXL2: ASXL transcriptional regulator 2 gene mutation; RUNX1-RUNX1T1: RUNX1-RUNX1 partner transcriptional co-repressor 1 genes fusion; BCR-ABL1: BCR-ABL1 fusion gene mutation; PTEN: phosphatase and tensin homolog gene mutation; CCND1: cyclin D1 gene mutation; CCND2: cyclin D2 gene mutation; CBL: casitas B-lineage lymphoma gene mutation; KRAS: Kirsten rat sarcoma virus gene mutation; FLT3: FMS-like tyrosine kinase 3 gene mutation; IDH2R140: isocitrate dehydrogenase 2 R140 gene mutation; CBFB-MYH11: core binding factor beta subunit-myosin heavy chain 11 genes fusion; PML-RARa: promyelocytic leukemia/retinoic acid receptor alpha gene mutation; ASXL1: ASXL transcriptional regulator 1 gene mutation; ZRSR2: zinc finger, RNA-binding motif and serine/arginine rich 2 gene mutation; BCORL1: BCORL1 gene mutation; BRCC3: BRCA1/BRCA2-containing complex 3 gene mutation. |
Relapse occurred in 134 patients (69.4%), with a significant difference between the inv(16) group (n = 61, 80.3%) and the t(8;21) group (n = 73, 62.4%) (P = 0.008). The percentage of deaths was 25.9%, and there was no difference between groups (P = 0.266).
The analysis of the characteristics of cases with a specific chromosomal alteration showed a significant difference in the OS of trisomy 22 (16.0 months, IQR: 1.7 - 28.0, P = 0.011) and loss of chromosome Y (48.0 months, IQR: 26.9 - 93.5, P = 0.004). There was no difference in the rest of the variables except for sex in loss of sex chromosomes (X and Y) and chromosome number in numerical chromosome abnormalities (Table 2).
Click to view | Table 2. Differences in the Characteristics of 193 CBF-AML Cases With Chromosome Aberrations |
In the entire sample, including patients with or without additional mutations, the median OS was 28.8 months (IQR: 12.0 - 60.0). In the inv(16) group, the median OS was 23.2 months (IQR: 10.0 - 50.6), and the 5-year OS was 53.7 (95% confidence intervals (CI): 40.0-72.0%). In the t(8;21) group, the median OS was 36.0 months (IQR: 16.8 - 60.0), and the 5-year OS was 73.4 (95% CI, 65.1-82.7%). There was no significant difference between the CBF-AML groups in the survival analysis (log-rank test, P = 0.062) (Fig. 2).
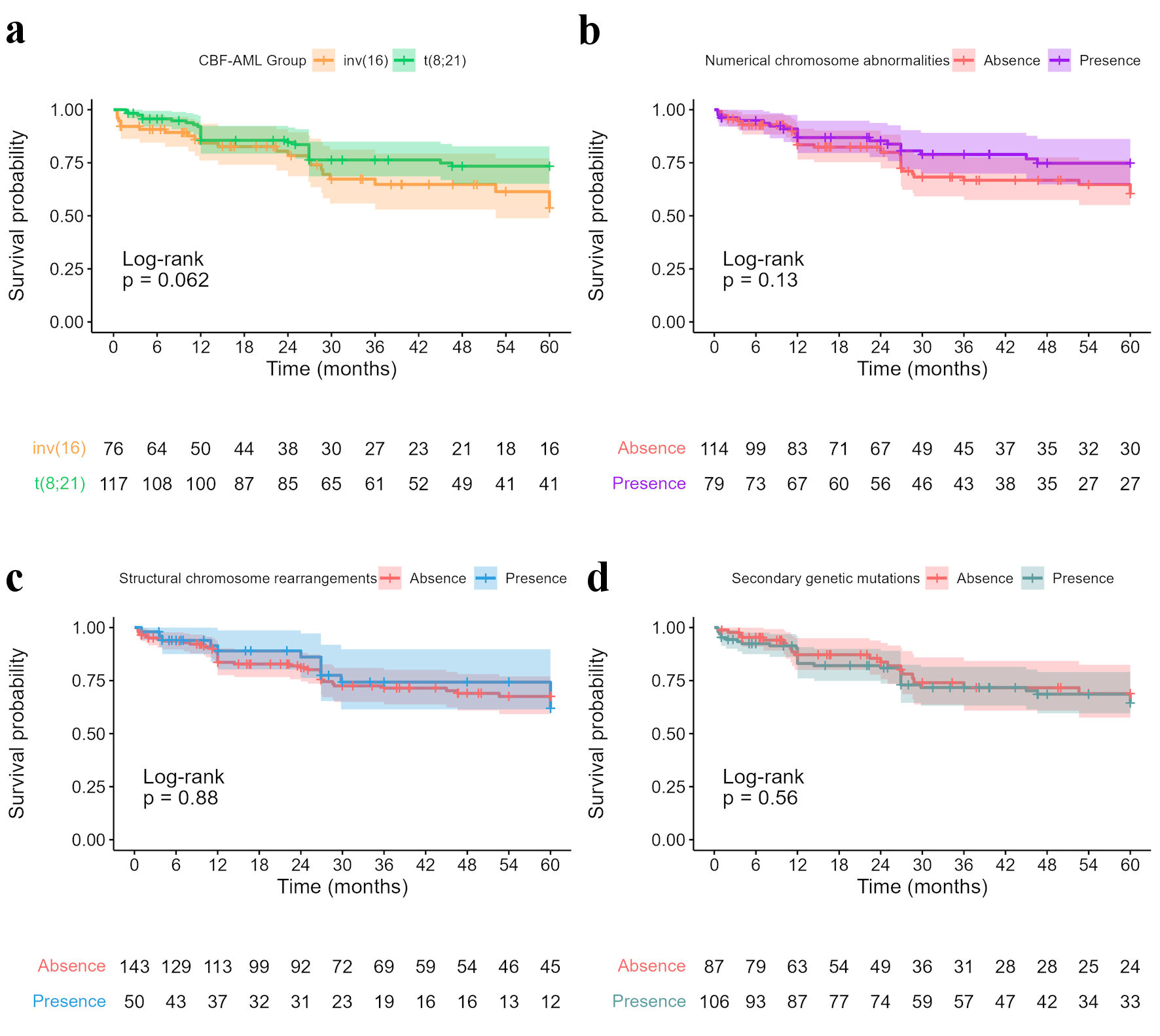 Click for large image | Figure 2. (a) Kaplan Meier curves for core binding factor acute myeloid leukemia (CBF-AML) groups showed no significant difference between groups. Five-year survival data of CBF-AML cases with secondary genetic findings: (b) Numerical chromosome abnormalities (5-year OS: 74.8%, 95% CI: 64.8 - 86.2, versus 60.4%, 95% CI: 50.1 - 72.9); (c) Structural chromosome rearrangements (5-year OS: 61.9%, 95% CI: 45.1 - 84.9, versus 67.5%, 95% CI: 59.2 - 77.0); (d) Secondary genetic mutations (5-year OS: 64.5%, 95% CI: 54.6 - 76.1, versus 68.9%, 95% CI: 57.5 - 82.4). CI: confidence interval; OS: overall survival. |
The 5-year OS rates with and without structural chromosome rearrangements were 61.9% (95% CI: 45.1 - 84.9) and 67.5% (95% CI: 59.2 - 77.0), respectively, with no significant difference (P = 0.88) and a hazard ratio (HR) of 0.95 (95% CI: 0.50 - 1.80; P = 0.884). Cases with numerical chromosome abnormalities had a 5-year OS of 74.8% (95% CI: 64.8 - 86.2), while those without chromosome abnormalities had a 5-year OS of 60.4% (95% CI: 50.1 - 72.9) (P = 0.13), with an HR of 0.64 (95% CI: 0.36 - 1.20; P = 0.139). The 5-year OS of patients with and without secondary genetic mutations was 64.5% (95% CI: 54.6 - 76.1) and 68.9% (95% CI: 57.5 - 82.4), respectively (P = 0.56), with an HR of 1.20 (95% CI: 0.67 - 2.10; P = 0.568) (Fig. 2). In addition, the presence of secondary genetic aberrations did not lead to a difference in the 5-year OS rate between the inv(16) and t(8;21) groups.
However, among individual survival analyses of secondary genetic aberrations, we found a difference in the 5-year OS long-rank test with the presence of 7q deletion (“del(7)”), trisomy 22 (“+22”) and NRAS mutations (Fig. 3). The 5-year OS for the presence of del(7) was significantly worse (27.8% versus 67.7%, P = 0.029) in terms of 5-year OS in the Cox univariate analysis (HR: 3.40; 95% CI: 1.00 - 11.00; P = 0.041). The 5-year OS of patients with and without trisomy 22 was 38.6% and 67.9%, respectively (P = 0.038), thus indicating a shorter OS in the presence of the aberration (HR: 2.8; 95% CI: 1.00 - 7.90; P = 0.049). In addition, we found that NRAS mutation had a worse 5-year OS in the t(8;21) group (25.0% versus 75.4%, PL = 0.014), with an HR of 4.00 (95% CI: 1.20 - 13.00; P = 0.025). However, when the total sample was included in this analysis, we found no significant difference.
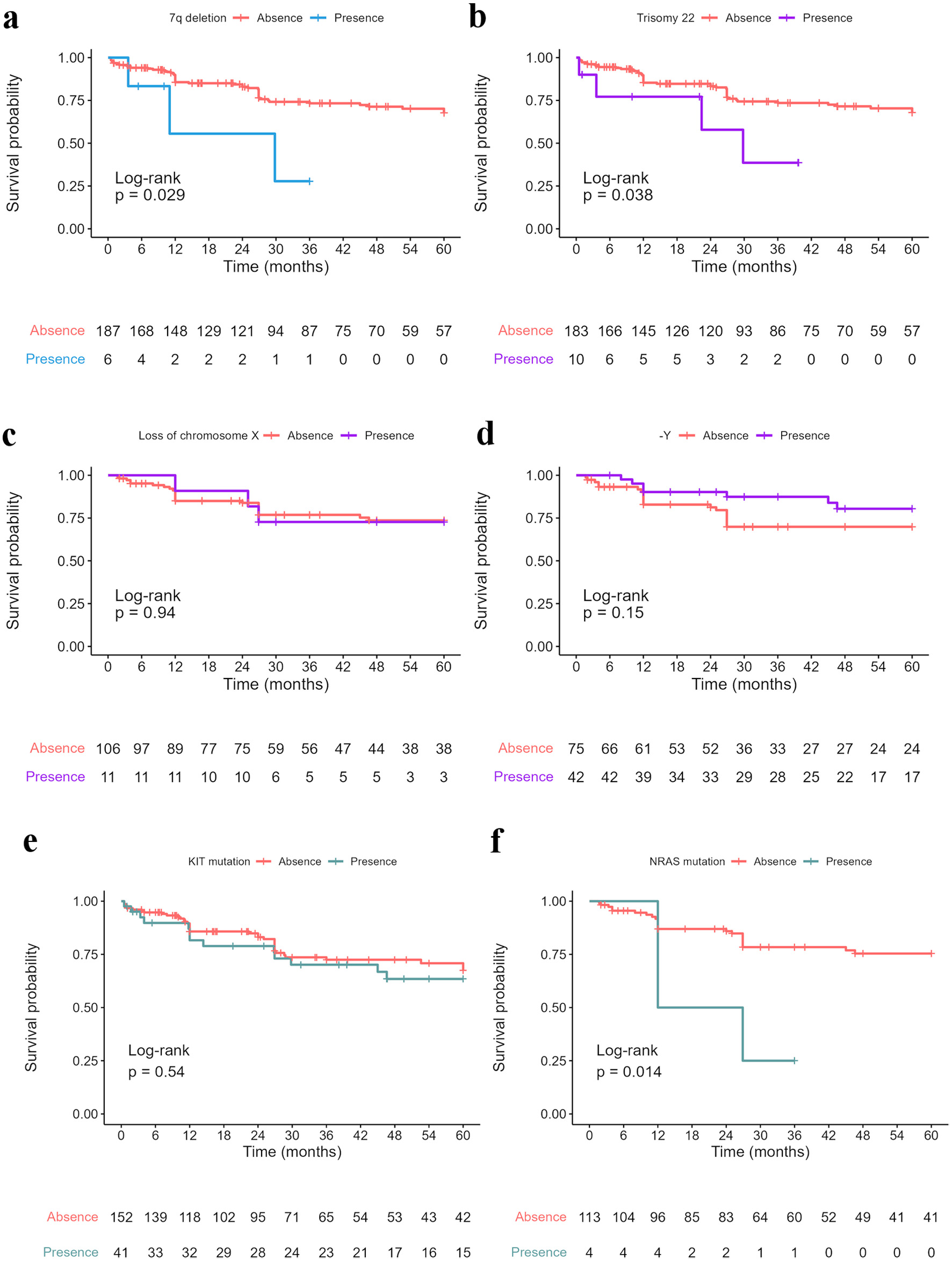 Click for large image | Figure 3. Five-year survival data of CBF-AML cases with specific secondary genetic aberration: (a) 7q deletion “del(7)” (5-year OS in total sample: 27.8%, 95% CI: 5.39 - 100, versus 67.7%, 95% CI: 60.1 - 76.2); (b) Trisomy 22 “+22” (5-year OS in total sample: 38.6%, 95% CI: 13.5 - 100, versus 67.9%, 95% CI: 60.2 - 76.5); (c) Loss of chromosome X “-X” (5-year OS in t(8;21) group: 72.7%, 95% CI: 50.6 - 100, versus 73.6%, 95% CI: 64.9 - 83.5); (d) Loss of chromosome Y “-Y” (5-year OS in t(8;21) group: 80.4%, 95% CI: 68.2 - 94.9, versus 69.9%, 95% CI: 59.6 - 82.0); (e) KIT mutation (5-year OS in total sample: 63.5%, 95% CI: 49.2 - 81.8, versus 67.4%, 95% CI: 58.7 - 77.4); (f) NRAS mutation (5-year OS in t(8;21) group: 25.0%, 95% CI: 4.58 - 100, versus 75.4%, 95% CI: 67.1 - 84.7). -Y: loss of chromosome Y; KIT: receptor tyrosine kinase gene KIT mutation; NRAS: neuroblastoma RAS viral gene mutation. CI: confidence interval; OS: overall survival. |
We performed multivariable analysis adjusted for sex, age group and relapse. These results were consistent with the univariate analysis except for NRAS mutation, which had no significant association (HR: 3.0; 95% CI: 0.86 - 10.2; P = 0.084), in contrast with univariate analysis (Table 3).
Click to view | Table 3. Univariate and Multivariate Cox Regression of Survival Outcomes in Patients With CBF-AML |
| Discussion | ▴Top |
Among 193 cases extracted from the Mitelman Database, we found that the incidence in t(8;21) was higher than in inv(16), with a ratio of approximately 1.5:1. This is consistent with previous studies where this rate is even higher in children, ranging from 2:1 to 4:1 [6, 12]. The proportions of structural and numerical chromosome rearrangements in our study were 25.9% and 40.9%, respectively, and the prevalence of secondary genetic mutations were 54.9% among individuals with CBF-AML. These additional aberrations were more frequent in the t(8;21) subgroup. A previous study hypothesized that RUNX1-RUNX1T1 requires a higher number of additional genetic aberrations to initiate leukemogenesis [13]. Additionally, there were certain chromosomal rearrangements that were predominant depending on each CBF-AML entity; t(9;22), del(7), trisomy 22 and trisomy 8 were more frequent in patients with inv(16), whereas del(9) and loss of sex chromosomes were more frequent in t(8;21). These results are consistent with previous studies [8, 10, 14].
Even though CBF-AML has a good prognosis, there is heterogeneity in the results of many studies with respect to the differences between inv(16)/CBFβ-MYH11 and t(8;21)/RUNX1-RUNXIT1 CBF-AML subtypes. A study in Italy showed a poorer prognosis with t(8;21) chromosomal rearrangement, and a Chinese study identified a trend toward worse OS in t(8;21) patients [15, 16]. We found that inv(16) 5-year OS was slightly better than t(8;21), but the survival analysis showed no significant difference between groups. Our results are consistent with the study of 91 Chinese children with newly diagnosed CBF-AML (5-year OS rate: 72% vs. 88%, P > 0.05) and a study with 537 CBF-AML patients from 12 institutions in the USA and Europe that found similar prognoses in the inv(16) and t(8;21) groups (5-year OS rate: 68% vs. 62%, P = 0.11) [10, 17].
The presence of structural chromosome rearrangements, numerical chromosome abnormalities and secondary genetic mutations were not associated with a significantly worse prognosis. However, we found some secondary genetic aberrations that led to significant differences in the prognosis. Previous studies found no significant impact of del(7) in CBF-AML patients [10, 17]; in contrast, our study found a worse prognosis in the presence of del(7) but no significant difference in the CBF-AML subgroup survival analysis. Trisomy 22 showed an adverse impact on the prognosis of patients when analyzed in the total sample but had no significant difference in the inv(16) group alone. This is consistent with a study that enrolled 370 patients with CBF-AML in the USA that found no significant difference in survival in the presence of trisomy 22 [18], but other studies have reported an association between this aberration and improved outcomes [19]. These findings suggest the need for further investigation to identify the role of del(7) and trisomy 22 in the prognosis of patients with CBF-AML.
Studies conducted in the USA and Europe reported a better OS in the presence of del(9) [10, 19]. Nevertheless, the prognostic impact of del(9) in our study showed no significant difference in the 5-year OS, which is similar to previous findings [18, 20]. There is no consensus on the impact of sex chromosome deletion on the prognosis of CBF-AML, specifically in patients with t(8;21), where the incidence is higher. The loss of the X chromosome is associated with a better prognosis in some studies [21-23], whereas our study found no significant difference in the 5-year OS, similar to the findings of Han et al [10]. In loss of the Y chromosome, our results showed a longer 5-year OS in the total sample but no difference in the t(8;21) subgroup survival analysis. A Chinese study found a high relapse risk and a shorter OS in these patients [24], but other studies showed no significant difference [25]. In fact, Liu et al found a higher 5-year OS rate of CBF-AML children with sex chromosome deletion but with no statistical significance [17], similar to our results but in the pediatric population. The association between trisomy 8 and prognosis has also not been well established. Some studies have found an association with a longer OS, an adverse impact or no significant difference in prognosis, as we did in our study [13, 20, 26].
KIT mutations have the highest incidence in CBF-AML patients and were the most common secondary genetic mutation in our study. Most previous studies showed that KIT mutations were associated with a shorter OS and a high recurrence rate [15, 25, 27]. However, our study did not show a significant impact in patients with KIT mutations in either inv(16) or t(8;21). These results are similar to the findings of Wu et al in the inv(16) group, but they contrast with the t(8;21) group, which is usually associated with poorer prognosis [15, 25]. NRAS mutations showed a significantly worse prognosis in the t(8;21) group, but previous evidence found an unclear role in the outcome. Some studies indicate that there is no impact on OS, and others show a better or worse prognosis [25, 26, 28]. Finally, we found no impact on prognosis with the presence of ASXL1, ASXL2, or FLT3 mutations, which is similar to other studies [15, 29, 30].
This study has some limitations. First, we exported the cases from a database of published papers, which means that our total number of cases could be affected by underreporting and can underestimate our results. Additionally, the year of publication could affect our results because the OS could improve by the development of new therapies. Another limitation was the diversity in the reporting of the data, which reduced our sample mainly because some studies did not report OS as an outcome or reported it in groups or in conjunction with other types of AML, which did not provide access to participant data. In cases with KIT or FLT3 mutations, we did not consider the subtypes because not all studies reported them. For the analysis, we only considered single aberrations and not whole karyotype abnormalities, which could underestimate or overestimate our conclusions. In addition, we found some mutations that were not very frequent, and several had one or two observations, which did not allow us to evaluate the differences in terms of OS in a significant way.
In conclusion, the use of a large and varied database such as Mitelman allowed our study to demonstrate that CBF-AML patients with inv(16) or t(8;21) have heterogeneous cytogenetic characteristics. However, this study found that patients with CBF-AML had a good prognosis but no difference in the 5-year OS between the inv(16) and t(8;21) groups. Our findings contribute to the knowledge about CBF-AML by highlighting that the presence of del(7), trisomy 22 and NRAS mutations showed a potential prognostic impact in CBF-AML patients. Further investigation with a larger sample is needed to clarify their role in the prognosis after excluding other secondary genetic aberrations and other factors.
| Supplementary Material | ▴Top |
Suppl 1. Flow diagram of the study participants.
Suppl 2. Glossary of variables.
Suppl 3. Structural chromosome rearrangements among 193 cases with CBF-AML from Mitelman Database.
Suppl 4. Numerical chromosome abnormalities among 193 cases with CBF-AML from Mitelman Database.
Suppl 5. Secondary genetic mutations among 193 cases with CBF-AML from Mitelman Database.
Acknowledgments
The authors wish to thank Dr. J. De la Cruz and Dr. C. Gutierrez for their suggestions for the statistical analysis, Dr. J. Faya for his help while programming in R and Dr. F. Romani for his critical review. And to our families who encouraged us in the development of this study
Financial Disclosure
The study is self-financed.
Conflict of Interest
The authors declare that this research was done with the purpose of obtaining the title of medical surgeon.
Informed Consent
Informed consent was not applicable for this study.
Author Contributions
R. Chapilliquen: conceptualization, methodology, software, validation, formal analysis, investigation, resources, data curation, writing-original draft, writing-review and editing, and visualization. M. Corbacho: conceptualization, methodology, validation, investigation, data curation, writing-original draft, writing-review and editing, and visualization. R. Zapata: conceptualization, validation, resources, supervision, and writing-review and editing.
Data Availability
The data analyzed in this study were obtained from The National Cancer Institute’s Mitelman Database of Chromosome Aberrations and Gene Fusions.
| References | ▴Top |
- Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7-33.
doi pubmed - Jung J, Cho BS, Kim HJ, Han E, Jang W, Han K, Lee JW, et al. Reclassification of acute myeloid leukemia according to the 2016 WHO Classification. Ann Lab Med. 2019;39(3):311-316.
doi pubmed pmc - Kuykendall A, Duployez N, Boissel N, Lancet JE, Welch JS. Acute myeloid leukemia: the good, the bad, and the ugly. Am Soc Clin Oncol Educ Book. 2018;38:555-573.
doi pubmed - Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer. 2002;2(7):502-513.
doi pubmed - Sangle NA, Perkins SL. Core-binding factor acute myeloid leukemia. Arch Pathol Lab Med. 2011;135(11):1504-1509.
doi pubmed - Faber ZJ, Chen X, Gedman AL, Boggs K, Cheng J, Ma J, Radtke I, et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat Genet. 2016;48(12):1551-1556.
doi pubmed pmc - Khan M, Cortes J, Qiao W, Alzubaidi MA, Pierce SA, Ravandi F, Kantarjian HM, et al. Outcomes of patients with relapsed core binding factor-positive acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2018;18(1):e19-e25.
doi pubmed pmc - Solh M, Yohe S, Weisdorf D, Ustun C. Core-binding factor acute myeloid leukemia: Heterogeneity, monitoring, and therapy. Am J Hematol. 2014;89(12):1121-1131.
doi pubmed - Sun A, Chao D, Chen S, Dai H, Depei W. The aberrations of cytogenetics and molecular genetics in core binding factor acute myeloid leukemia. Blood [Internet]. 2015;126(23):4802-4802.
- Han SY, Mrozek K, Voutsinas J, Wu Q, Morgan EA, Vestergaard H, Ohgami R, et al. Secondary cytogenetic abnormalities in core-binding factor AML harboring inv(16) vs t(8;21). Blood Adv. 2021;5(10):2481-2489.
doi pubmed pmc - Paschka P, Marcucci G, Ruppert AS, Mrozek K, Chen H, Kittles RA, Vukosavljevic T, et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol. 2006;24(24):3904-3911.
doi pubmed - Li W, Mi YC, Liu BC, Zhou CL, Lin D, Wang HJ, Liu XP, et al. [Clinical and cytogenetic features and their influencing factors of core binding factor acute myeloid leukemia]. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2011;33(5):517-524.
pubmed - Jahn N, Terzer T, Strang E, Dolnik A, Cocciardi S, Panina E, Corbacioglu A, et al. Genomic heterogeneity in core-binding factor acute myeloid leukemia and its clinical implication. Blood Adv. 2020;4(24):6342-6352.
doi pubmed pmc - Hsiao HH, Liu YC, Wang HC, Tsai YF, Wu CH, Cho SF, Hsu JF, et al. Additional chromosomal abnormalities in core-binding factor acute myeloid leukemia. Genet Mol Res. 2015;14(4):17028-17033.
doi pubmed - Wu TM, Xue SL, Li Z, Yu JQ, Wang J, Wang BR, Wan CL, et al. [Prognostic value of KIT and other clonal genetic mutations in core-binding factor acute myeloid leukemia]. Zhonghua Xue Ye Xue Za Zhi. 2021;42(8):646-653.
doi pubmed pmc - Mosna F, Papayannidis C, Martinelli G, Di Bona E, Bonalumi A, Tecchio C, Candoni A, et al. Complex karyotype, older age, and reduced first-line dose intensity determine poor survival in core binding factor acute myeloid leukemia patients with long-term follow-up. Am J Hematol. 2015;90(6):515-523.
doi pubmed - Liu C, Chen XY, Yi MH, Wu WQ, Ruan M, Zhu XF. [Clinical features and prognosis of core binding factor acute myeloid leukemia in children]. Zhongguo Dang Dai Er Ke Za Zhi. 2020;22(7):739-743.
doi pubmed pmc - Appelbaum FR, Kopecky KJ, Tallman MS, Slovak ML, Gundacker HM, Kim HT, Dewald GW, et al. The clinical spectrum of adult acute myeloid leukaemia associated with core binding factor translocations. Br J Haematol. 2006;135(2):165-173.
doi pubmed - Marcucci G, Mrozek K, Ruppert AS, Maharry K, Kolitz JE, Moore JO, Mayer RJ, et al. Prognostic factors and outcome of core binding factor acute myeloid leukemia patients with t(8;21) differ from those of patients with inv(16): a Cancer and Leukemia Group B study. J Clin Oncol. 2005;23(24):5705-5717.
doi pubmed - Schlenk RF, Benner A, Krauter J, Buchner T, Sauerland C, Ehninger G, Schaich M, et al. Individual patient data-based meta-analysis of patients aged 16 to 60 years with core binding factor acute myeloid leukemia: a survey of the German Acute Myeloid Leukemia Intergroup. J Clin Oncol. 2004;22(18):3741-3750.
doi pubmed - Klein K, Kaspers G, Harrison CJ, Beverloo HB, Reedijk A, Bongers M, Cloos J, et al. Clinical impact of additional cytogenetic aberrations, ckit and ras mutations, and treatment elements in pediatric t(8;21)-AML: results from an international retrospective study by the International Berlin-Frankfurt-Munster Study Group. J Clin Oncol. 2015;33(36):4247-4258.
doi pubmed pmc - Yue-Ping J, Ying-Xi Z, Ai-Dong L, Le-Ping Z, Gui-Lan L. Prognostic significance of M2 sex chromosome deletion in childhood acute myeloid leukemia. Chinese Journal of Contemporary Pediatrics [Internet]. 2015;17(2):168-171.
- Chen G, Zhou W, Gong D, Li Y, Huang S, Wang N, Xu Q, et al. Loss of X chromosome predicts favorable prognosis in female patients with t(8;21) acute myeloid leukemia. Leuk Lymphoma. 2020;61(5):1168-1177.
doi pubmed - Zhou W, Chen G, Gong D, Li Y, Huang S, Wang N, Xu Q, et al. Loss of the Y chromosome predicts a high relapse risk in younger adult male patients with t(8;21) acute myeloid leukemia on high-dose cytarabine consolidation therapy: a retrospective multicenter study. Leuk Lymphoma. 2020;61(4):820-830.
doi pubmed - Ishikawa Y, Kawashima N, Atsuta Y, Sugiura I, Sawa M, Dobashi N, Yokoyama H, et al. Prospective evaluation of prognostic impact of KIT mutations on acute myeloid leukemia with RUNX1-RUNX1T1 and CBFB-MYH11. Blood Adv. 2020;4(1):66-75.
doi pubmed pmc - Rogers HJ, Wang X, Xie Y, Davis AR, Thakral B, Wang SA, Borthakur G, et al. Comparison of therapy-related and de novo core binding factor acute myeloid leukemia: A bone marrow pathology group study. Am J Hematol. 2020;95(7):799-808.
doi pubmed - Cairoli R, Beghini A, Grillo G, Nadali G, Elice F, Ripamonti CB, Colapietro P, et al. Prognostic impact of c-KIT mutations in core binding factor leukemias: an Italian retrospective study. Blood. 2006;107(9):3463-3468.
doi pubmed - Boissel N, Leroy H, Brethon B, Philippe N, de Botton S, Auvrignon A, Raffoux E, et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia. 2006;20(6):965-970.
doi pubmed - Paschka P, Schlenk RF, Gaidzik VI, Herzig JK, Aulitzky T, Bullinger L, Spath D, et al. ASXL1 mutations in younger adult patients with acute myeloid leukemia: a study by the German-Austrian Acute Myeloid Leukemia Study Group. Haematologica. 2015;100(3):324-330.
doi pubmed pmc - Kawashima N, Akashi A, Nagata Y, Kihara R, Ishikawa Y, Asou N, Ohtake S, et al. Clinical significance of ASXL2 and ZBTB7A mutations and C-terminally truncated RUNX1-RUNX1T1 expression in AML patients with t(8;21) enrolled in the JALSG AML201 study. Ann Hematol. 2019;98(1):83-91.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
World Journal of Oncology is published by Elmer Press Inc.